Lehre - Supramolekulare Chemie - Supramolekulare Katalyse
Einleitung
Supramolekulare Katalyse beinhaltet die Schritte Erkennung und Transformation, d.h. ein supramolekularer Katalysator bewirkt nicht nur die Beschleunigung einer Reaktion, er sollte darüber hinaus - wie ein Enzym - aus einer Vielzahl strukturell ähnlicher Substrate selektiv nur eines umsetzen. Schließlich muss das Produkt der Reaktion vom Katalysator weniger gut gebunden werden als das Edukt, damit es zu einem echten Turnover kommt. Tatsächlich stellt diese Produktinhibition bei vielen supramolekularen Katalysatoren ein großes Problem dar.
Die enzymatische Katalyse beruht in vielen Fällen darauf, dass das Enzym den Übergangszustand einer Reaktion besser bindet als die Edukte und das Produkt. Dieses Prinzip ist bisher in wenigen supramolekularen Katalysatoren verwirklicht.
Häufig werden beim Design supramolekularer Katalysatoren reaktive Gruppen, die die gewünschte Reaktion katalysieren, in räumlicher Nähe zum gebundenen Substrat platziert. Diese Nähe zum Reaktionszentrum soll ausreichen, um die Katalyse zu bewirken und Produktinhibition soll nur in untergeordnetem Maße stattfinden.
Auf kombinatorische Verfahren zur Katalysatorentwicklung wurde im vorigen Kapitel hingewiesen.
Eine Reihe verschiedener Reaktionstypen können durch Katalysatoren beschleunigt werden.
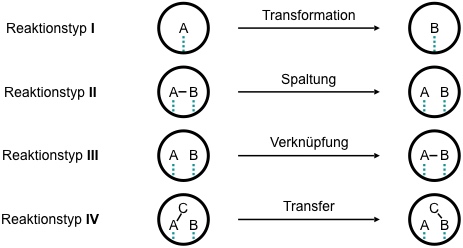
Während Typ I und IV entropisch neutral sind, ist II entropisch günstig und Typ III entropisch ungünstig. Daher ist es oft erfolgversprechender, Katalysatoren für Reaktionen zu entwickeln, die Typ II entsprechen, z.B. für Hydrolysen.
Hydrolase Mimetika
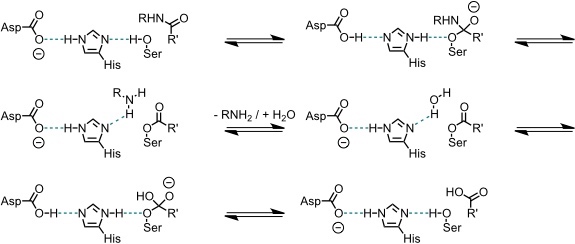
Grundlegende Arbeiten im Bereich der supramolekularen Katalyse beschäftigten sich in der Gruppe um D. J. Cram mit der Entwicklung eines Mimetikums für das Enzym Chymotrypsin, welches Peptidbindungen spaltet. Dieses Enzym enthält im aktiven Zentrum ein sogenanntes charge-relay System aus einer Aspartat/Histidin/Serin-Triade. Durch dieses gekoppelte System wird die Nucleophilie der Serin OH-Gruppe erhöht.

Bei der eigentlichen Reaktion wird die Peptidbindung unter Acylierung der Serin OH-Gruppe gespalten. In einem zweiten Schritt wird das acylierte Serin dann durch Wasser hydrolysiert.
Das von Cram entworfene potentielle Chymotrypsin Mimetikum besitzt folgende Struktur.
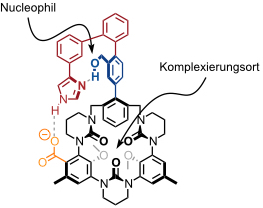
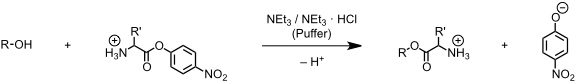
An diesen Rezeptor näherte man sich schrittweise an. Zunächst wurde das schwarz dargestellte Derivat synthetisiert. Diese Verbindung bindet protonierte Aminosäure-p-nitrophenylester, wobei bei der Komplexbildung die OH-Gruppe in räumlicher Nähe zur Estergruppe des Substrats angeordnet wird. Die Acylierung der OH-Gruppe wird dadurch 1011 mal schneller als die der OH-Gruppe von 3-Phenylbenzylalkohol, einer Modellverbindung, der die makrocyclische Rezeptoreinheit fehlt.
Nun wurde ein weiteres Derivat des Rezeptors (in einer 30 stufigen Synthese!) dargestellt, das sich vom geplanten Katalysator nur noch durch das Fehlen der Carboxylgruppe unterschied. Dieses wird praktisch augenblicklich durch p- Nitrophenylester von Aminosäuren acyliert, allerdings an der Imidazolgruppe. Erst eine langsame Umlagerung ergibt anschließend das O-acylierte Produkt.
Der Rezeptor mit der Carboxylgruppe ist nicht mehr beschrieben worden.
Diese Untersuchungen zeigen, dass es zwar möglich ist, durch Imitation von natürlichen Wirkungsprinzipien supramolekulare Systeme zu erhalten, die charakteristische Eigenschaften von Katalysatoren besitzen, dass sich aber auch bei sorgfältiger Planung der Strukturen unerwartete Ergebnisse ergeben können. Man ist daher davon abgekommen, Katalysatoren durch vielstufige Synthesen darzustellen, sondern man versucht heute eher, bekannte Rezeptoren als Basis für supramolekulare Katalysatoren zu verwenden, welche mit geeigneten reaktiven Gruppen versehen werden.
Das charge-relay System des Chymotrypsins stellt aber nach wie vor eine Grundlage für neuere Entwicklungen dar, wie das folgende Beispiel zeigt, bei dem ein modifiziertes Cyclodextrin, eine Glutamat- und eine Histidineinheit in strategischen Positionen an eine α-helikale Oligopeptidkette gebunden wurden.
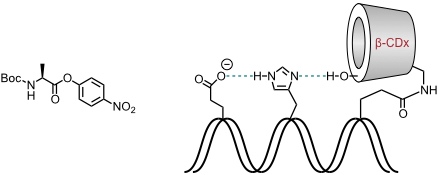
[Quellen: Tsutsumi, H.; Hamasaki, K.; Mihara, H.; Ueno, A. Bioorg. Med. Chem. Lett. 2000, 10, 741-743: <doi>. Tsutsumi, H.; Hamasaki, K.; Mihara, H.; Ueno, A. J. Chem. Soc., Perkin Trans. 2 2000, 1813-1818: <doi>.]
Cyclodextrine
Eine wichtige Rolle in der Entwicklung supramolekularer Katalysatoren spielen Cyclodextrinderivate. Sie sind aus verschiedenen Gründen als Enzymmodelle interessant:
- sie sind wasserlöslich,
- Gäste werden im Hohlraum reversibel gebunden, allerdings verläuft die Freisetzung der Gäste meist nicht so schnell wie bei Enzymen,
- aufgrund ihrer Chiralität ist prinzipiell eine enantioselektive Katalyse denkbar.
Schon unmodifizierte Cyclodextrine beschleunigt die Hydrolyse von Carbonsäure- und Phosphatestern. Der Mechanismus der Hydrolyse ähnelt dabei dem des Chymotrypsins, auch wenn kein Imidazolrest beteiligt ist.
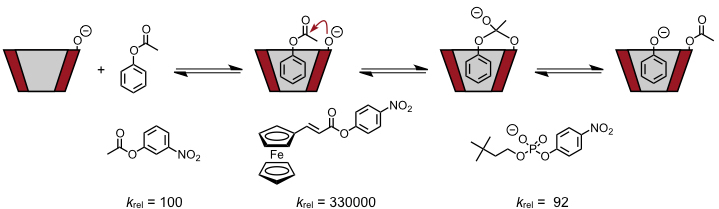
- Das Cyclodextrin wirkt nicht als Katalysator, da es nicht zu Turnover kommt.
- Die Reaktion ähnelt dem ersten Schritt der Esterhydrolyse einer Serinprotease.
- Die Reaktionsgeschwindigkeit hängt von der Stabilität des Komplexes und der Orientierung der zu hydrolysierenden Estergruppe zum angreifenden Nucleophil (deprotonierte 2-OH-Gruppe) ab.
Werden Cyclodextrine mit geeigneten Substituenten versehen, kann man zu einer Reihe interessanter Katalysatoren gelangen. Vor allem die Gruppe um R. Breslow hat sich intensiv mit der Entwicklung solcher Systeme beschäftigt.
Esterase Mimetikum
Eine Esteraseaktivität wurde z.B. für folgendes dimere Cyclodextrin beobachtet.
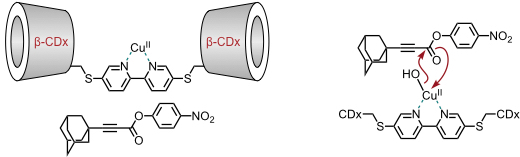
Diese Verbindung katalysiert die Spaltung geeigneter Ester mit hoher Effizienz, wobei die Aktivität durch die Einlagerung der hydrophoben Substituenten in die Hohlräume der beiden Cyclodextrinuntereinheiten zu erklären ist. Dadurch gelangt das Kupferion in die Nähe der Estergruppe und kann deren Spaltung auslösen.
Der Vorteil dieses Katalysators besteht darin, dass die einzelnen Fragmente des Substrats mit sehr viel geringerer Affinität gebunden werden als das Substrat selber, sodass keine Produktinhibition beobachtet wird und echter Turnover möglich ist.
[Quelle: Breslow, R.; Zhang, B. J. Am. Chem. Soc. 1992, 114, 5882-5883: <doi>.]
Ribonuclease Mimetikum
Ribonucleaseaktivität wurde für ein β-Cyclodextrinderivat mit zwei Imidazolresten nachgewiesen. Wie im natürlichen Vorbild ist für die katalytische Aktivität ein Imidazol- und ein Imidazoliumrestes verantwortlich. Dabei besitzt ein Derivat mit den Substituenten an benachbarten Glucoseeinheiten (AB) eine größere Aktivität als Derivate, in denen die Substituenten weiter voneinander entfernt sind (AC, AD).
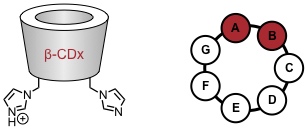
Die Regioselektivität des Cyclodextrins bei der Spaltung eines cyclischen Phosphatesters steht im Einklang mit der Wirkungsweise des natürlichen Katalysators.
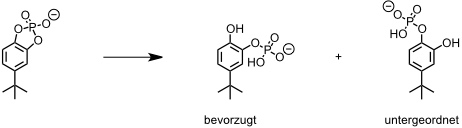
Dieses Ergebnis ist darauf zurückzuführen, dass sich durch die Geometrie der Einlagerung und durch die Stellung der Imidazolgruppen im gebildeten Zwischenprodukt die P-O–Bindung und die zur tert-Butylgruppe para-ständige P–O Bindung in den apicalen Positionen des fünffach koordinierten Phosphors befinden, während die restlichen P–O Bindungen in äquatorialen Positionen liegen. Nur apicale Substituenten können allerdings als Abgangsgruppen fungieren, sodass die bevorzugte Bildung des meta-Phosphatesters erklärbar ist.
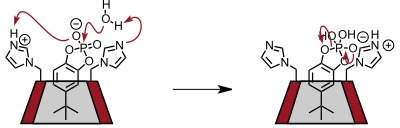
Eine Pseudororation, bei der apicale und äquatoriale Substituenten ineinander übergehen ist so langsam, dass der isomere para-Phosphatester nur bis zu 2% gebildet wird.
[Quelle: Breslow, R.; Schmuck, C. J. Am. Chem. Soc. 1996, 118, 6601-6605: <doi>.]
Andere Ribonuclease-Mimetika enthalten oft Zink(II) im katalytisch wirksamen Zentrum. Als Beispiel seien Calixarenderivate genannt, die in der Gruppe von D. N. Reinhoudt entwickelt worden sind.
Transaminase Mimetikum
Ein β-Cyclodextrinderivat mit einem Pyridoxalsubstituenten beschleunigt die Transaminierung von Ketosäuren.
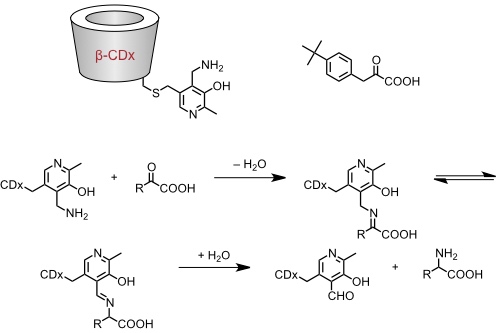
Eine Enantioselektivität wird bei dieser Umsetzung praktisch nicht beobachtet.
[Quelle: Breslow, R.; Hammond, M.; Lauer, M. J. Am. Chem. Soc. 1980, 102, 421-422: <doi>.]
Transketolase Mimetiku
Mit einem γ-Cyclodextrinderivat, das einen Thiazoliumsubstituenten trägt, kann die Benzoinkondensation katalysiert werden. Dabei ist der γ-Cyclodextrinhohlraum so groß, dass er zwei Moleküle des Substrats einschließen kann.
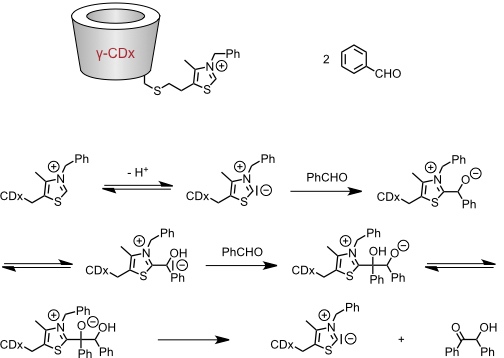
[Quelle: Breslow, R.; Kool, E. Tetrahedron Lett. 1988, 29, 1635-1638: <doi>.]
Polyazamakrocyclen
Phosphorylase Mimetikum
Ein einfacher Polyazamakrocyclus katalysiert die Dephosphorylierung von ATP zu ADP.
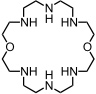
Die Reaktionsbeschleunigung beträgt das ca. 100fache über einen relativ weiten pH Bereich. Außerdem wird eindeutig Turnover beobachtet, da das Reaktionsprodukt ADP mit wesentlich geringerer Affinität gebunden wird als ATP.
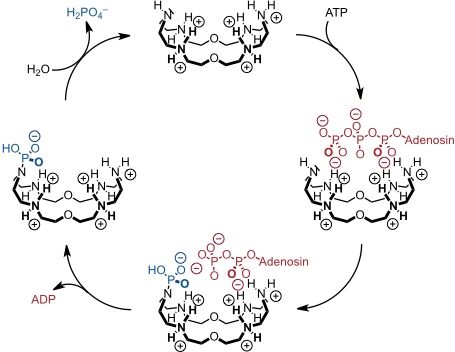
Als Intermediat tritt im katalytischen Zyklus ein Phosphoramidat auf. Dieses Intermediat kann auch als Phosphorylierungsreagenz verwendet werden. So kann es in Gegenwart von Mg2+ und Ca2+ Ionen ADP zu ATP umsetzen (ATP-Synthase).
[Quelle: Hosseini, M. W. "Supramolecular Catalysis of Phosphoryl Anion Transfer Processes" in: Supramolecular Chemistry of Anions, Bianchi, A.; Bowman-James, K.; Garcia-España, E. (Hrsg.), Wiley: New York, 1997, 421-448.
Cyclophane
Oxidase Mimetikum
In dem von F. Diederich entwickelten Cyclophan vermittelt der Thiazoliumring die Oxidation von einem Aldehyd zu einem Methylester.
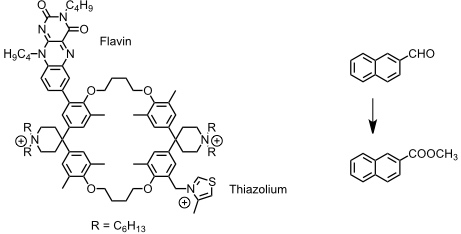
Das Cyclophan bindet den aromatischen Rest des Aldehyds in seinem Hohlraum. Es bildet sich ein Intermediat mit dem Thiazoliumring und anschließend erfolgt Hydridtransfer auf das Flavin. Durch Solvolyse mit Methanol wird das Produkt freigesetzt. Turnover erhält man durch elektrochemische Regenerierung des Flavins.
[Quelle: Mattei, P.; Diederich, F. Helv. Chim. Acta 1997, 80, 1555-1588: <doi>.]
Oxidase Mimetika
Eine regioselektive Oxidation von CH-Bindungen wurde von R. Breslow mit Hilfe eines Porphyrinkomplexes realisiert, der vier β-Cyclodextrinsubstituenten trägt.
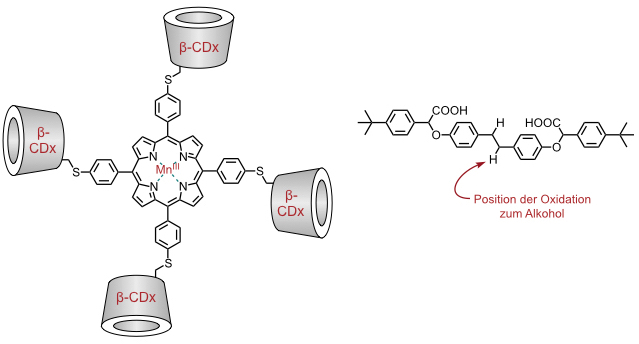
Die Regioselektivität der Oxidation resultiert aus der Einlagerung des Substrats in zwei Cyclodextrinringe des Katalysators und die sich daraus ergebende räumliche Nähe des Metallzentrums zur CH-Bindung. Die Carboxylatgruppen im Substrat dienen zur Erhöhung der Wasserlöslichkeit.
[Quelle: Breslow, R.; Zhang, X.; Huang, Y. J. Am. Chem. Soc. 1997, 119, 4535-4536: <doi>.]
Mimetika für Carotinmonooxygenasen, welche ebenfalls aus mit Cyclodextrinsubstituenten versehenen Porphyrinderivaten bestehen, wurden in der Gruppe von W.-D. Woggon entwickelt.
Koordinationskäfige
Die in der Gruppe von Fujita entwickelten Koordinationskäfige beeinflussen Diels-Alder Reaktionen.
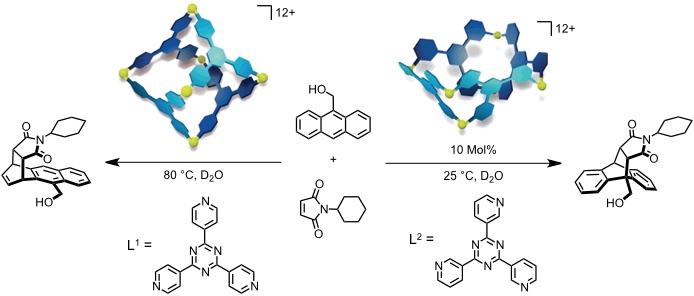
Der oktaedrische [Pd6L1]12+ Komplex bildet einen ternären Komplex mit N-Cyclohexylmaleimid und 6-Hydroxymethylanthacen. Unterhalb von 80 °C wird keine Reaktion beobachtet aber oberhalb von 80 °C kommt es zur regioselektiven Bildung eines Produktes, das außerhalb des Käfigs nicht gebildet wird. Dieses Produkt verbleibt im Käfig, Turnover wird also nicht beobachtet.
Katalytische Mengen des schalenförmigen [Pd6L2]12+ Komplexes führen zu einer signifikanten Beschleunigung derselben Diels-Alder Reaktion (99% Umsatz innerhalb von 5 h, 3% Umsatz unter denselben Bedingungen ohne Komplex). Turnover wird durch die leicht zugängliche Rezeptorkavität erklärt, die einen leichten Austausch des Produkts und der Reaktionspartner erlaubt.
[Quelle: Yoshizawa, M.; Tamura, M.; Fujita, M. Science 2006, 312, 251-254: <doi>.]
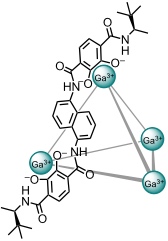
Der von Raymond beschriebene Koordinationstetraeder bindet kationische Verbindungen. Entsprechend beschleunigt er Reaktionen mit kationischen Zwischenprodukten oder Übergangszuständen, z.B. Aza-Cope Umlagerungen.
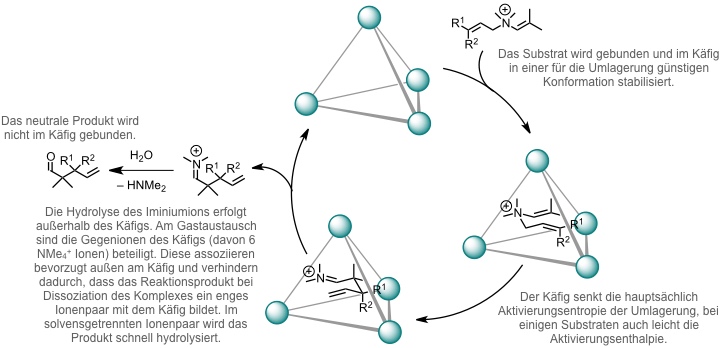
Für das Substrat mit R1 = H und R2 = iPr beträgt die Reaktionsbeschleunigung kcat / kuncat ca. 850 (50 °C, D2O/ Phosphatpuffer, pD 8,0). Mit katalytischen Mengen des Käfigs beobachtet man Turnover.
[Quelle: Fiedler, D.; van Halbeek, H.; Bergman, R. G.; Raymond, K. N. J. Am. Chem. Soc. 2006, 128, 10240-10252: <doi>.]
Derselbe Koordinationskäfig beschleunigt die sauer katalysierte Hydrolyse von Orthoformiaten bei pH 11 (!!). Die Ursache ist wiederum die Stabilisierung der positiv geladenen Zwischenstufen im Inneren des Käfigs und die damit verbundene Erhöhung der Basizität protonierbarer Spezies.
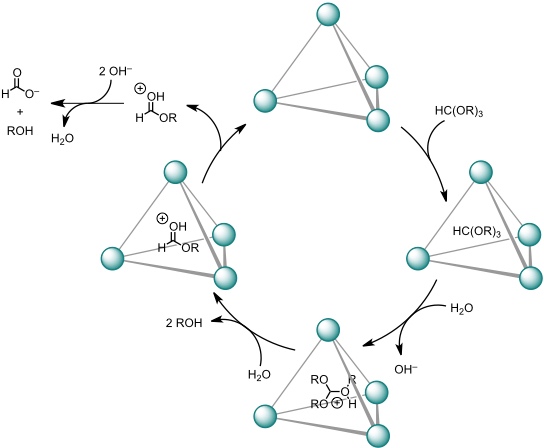
Die höchste Reaktionsbeschleunigung (kcat / kuncat = 3900) wurde für Tri-n-propylorthoformiat beobachtet (50 °C, H2O, pH 11,0). Der Zusatz von Triethylammoniumsalzen führt zu einer Inhibition der Reaktion, da deren Kationen mit hoher Affinität von dem Käfig gebunden werden.
[Quelle: Pluth, M. D.; Bergman, R: G.; Raymond, K. N. Science 2007, 316, 85-88: <doi>.]
Eine noch größere Reaktionsbeschleunigung um das 2,1 millionenfache wurde bei der Nazarov Cyclisierung des Pentamethylpentadienylkations, einer elektrocyclischen Ringschlussreaktion, beobachtet.
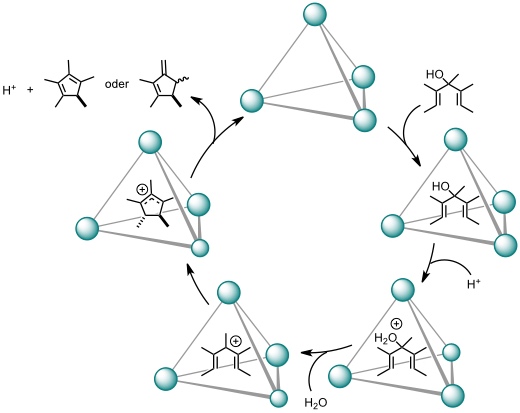
[Quelle: Hastings, C. J.; Pluth, M. D.; Bergman, R: G.; Raymond, K. N. J. Am. Chem. Soc. 2010, 132, 6938-6940: <doi>.]
Durch die Komplexbildung kann die Stereochemie einer Reaktion geändert werden. Bei der Solvolyse eines enantiomerenreinen Trichloracetimidats beobachtet man in Gegenwart einer racemischen Säure die erwartete Inversion des stereogenen Zentrums. In Anwesenheit eines (aufgrund der chiralen Substituenten) enantiomerenreinen Δ,Δ,Δ,Δ-konfigurierten Koordinationskäfigs beobachtet man überraschenderweise im großem Umfang die Retention des Stereozentrums. Analog reagiert auch (1-Chlorethyl)benzol.
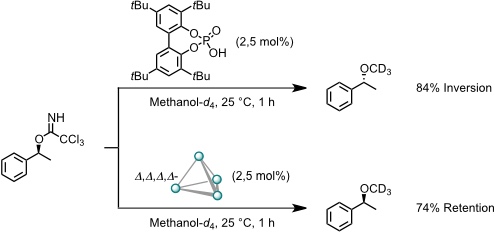
Man nimmt an, dass die Reaktion im Käfig über Carbeniumionen verlaufen, welche durch die Komplexbildung stabilisiert werden. Die Stereoselektivität der Weiterreaktion mit dem Lösungsmittel wird durch sterische Effekte kontrolliert, welches scheinbar nur von der Seite angelagert werden kann, auf der sich ursprünglich die Abgangsgruppe befand. Die Stereochemie der Kapsel hat keinen signifikanten Einfluss auf die Stereoselektivität der Reaktion.
[Quelle: Zhao, C.; Toste, F. D.; Raymond, K. N.; Bergman, R. G. J. Am. Chem. Soc. 2014, 136, 14409-14412: <doi>.]
Literatur
- Breslow, R. "Biomimetic Chemistry and Artificial Enzymes: Catalysis by Design" Acc. Chem. Res. 1995, 28, 146-153: <doi>.
- Murakami, Y.; Kikuchi, J.-I.; Hisaeda, Y.; Hayashida, O. "Artificial Enzymes" Chem. Rev. 1996, 96, 721-756: <doi>.
- Sanders, J. K. M. "Supramolecular Catalysis in Transition" Chem. Eur. J. 1998, 4, 1378-1383: <doi>.
- Breslow, R.; Dong, S. D. "Biomimetic Reactions Catalyzed by Cyclodextrins and Their Derivatives" Chem. Rev. 1998, 98, 1997-2011: <doi>.
- Molenveld, P.; Stikvoort, W. M. G.; Kooijman, H.; Spek, A. L.; Engbersen, J. F. J.; Reinhoudt, D. N. "Dinuclear and Trinuclear Zn(II) Calix[4]arene Complexes as Models for Hydrolytic Metallo-Enzymes. Synthesis and Catalytic Activity in Phosphate Diester Transesterification" J. Org. Chem. 1999, 24, 3896-3906: <doi>.
- Woggon, W.-D. "Metalloporphyrins as Active Site Analogues - Lessons from Enzymes and Ezyme Models" Acc. Chem. Res. 2005, 38, 127-136: <doi>.
- Pluth, M. D.; Bergman, R. G.; Raymond, K. N. "Proton-Mediated Chemistry and Catalysis in a Self-Assembled Supramolecular Host" Acc. Chem. Res. 2009, 42, 1650-1659: <doi>.
- Yoshizawa, M.; Klosterman, J. K.; Fujita, M. "Funktionale molekulare Reaktionskolben: neuartige Eigenschaften und Reaktionen in diskreten, selbstorganisierten Wirtmolekülen" Angew. Chem. 2009, 121, 3470-3490: <doi>.
- Brown, C. J.; Toste, F. D.; Bergman, R: G.; Raymond, K. N. "Supramolecular Catalysis in Metal-Ligand Cluster Hosts" Chem. Rev. 2015, 115, 3012-3035: <doi>.
- Nachtsheim, B. J. "Synthesen in Kavitäten" Nachr. Chem. 2015, 63,783-787: <doi>.
Letzte Änderung: 23-03-30. Email