Lehre - Spektroskopische Strukturaufklärung organischer Verbindungen - NMR-Spektroskopie
Physikalische Grundlagen
Die meisten Atomkerne besitzen einen Kern- oder Eigendrehimpuls P. In der klassischen Vorstellung ist dieser auf die Rotation der Kerne um eine Kernachse zurückzuführen. Wie viele andere atomare Größen auch, ist der Kerndrehimpuls gequantelt.
P = (h/2π) · [I(I + 1)]½
h = Plancksches Wirkungsquantum
I = Kernspinquantenzahl
Die Kernspinquantenzahl kann die Werte 0, ½, 1, 3/2, 2 ... 6 annehmen. Weder die Werte für I noch die für P lassen sich für die einzelnen Kernsorten vorhersagen.
Da mit einer rotierenden Ladung immer auch die Erzeugung eines Magnetfeldes verbunden ist, besitzt jeder Atomkern mit einer Kernspinquantenzahl ungleich Null ein magnetisches Moment.
μ = γ · P = γ · (h/2π) · [I(I + 1)]½
γ = gyromagnetisches oder magnetogyrisches Verhältnis
Das gyromagnetische Verhältnis beschreibt die Nachweisempfindlichkeit eines Kerns in einem NMR Experiment. Für jede Kernsorte mit I > 0 besitzt γ einen charakteristischen Wert. Kerne mit großem γ sind leichter zu beobachten (empfindlicher) als solche mit kleinem γ (unempfindlicher). Zwei der wichtigsten Bestandteile organischer Verbindungen, nämlich die Isotope 12C und 16O, besitzen ein I von 0. Diese Kerne sind daher NMR-spektroskopisch nicht nachweisbar. Andererseits ist das magnetische Moment der Kerne 1H (I = ½, γ = 26.7519 · 107 rad T-1 s-1) und 13C (I = ½, γ = 6.7283 · 107 rad T-1 s-1) ungleich Null. Die 1H- und 13C-NMR-Spektroskopie haben daher zentrale Bedeutung in der organischen Strukturanalyse. Weitere NMR-aktive Kerne sind z.B. 2H (I = 1), 19F (I = ½) und 31P (I = ½).
Bei Anlegen eines äußeren Magnetfeldes B0 kommt es zur sogenannten Richtungsquantelung, d.h. ein Kern, dessen magnetisches Moment in Feldrichtung μz parallel zum äußeren Magnetfeldes angeordnet ist, ist energieärmer als ein Kern, dessen magnetisches Moment dem äußeren Feld entgegengerichtet ist. Zur Beschreibung der Komponente Pz des Drehimpulses in Feldrichtung wird die magnetische Quantenzahl m benötigt.
Pz = m · (h/2π)
m = I, I - 1, I - 2, ..., -I
Für das magnetische Moment in Feldrichtung ergibt sich entsprechend.
μz = m · γ · (h/2π)
Kerne mit einem I von ½ haben also zwei Einstellungsmöglichkeiten in einem Magnetfeld (m = -½ und m = +½) und solche mit einem I von 1 haben drei (m = -1, m = 0, m = 1).
Die Energie eines magnetischen Dipols in einem Magnetfeld der magnetischen Flussdichte B0 beträgt:
E = -μz · B0 = -m · γ · (h/2π) · B0
Damit ergeben sich für einen Kern mit I = ½ zwei Energieniveaus, die sogenannten Kern-Zeeman-Niveaus, wobei der Spinquantenzahl m = +½ die energetisch günstigere Anordnung zugeordnet ist. Dieses Energieniveau wird quantenmechanisch durch die Spinfunktion α beschrieben, während die energetisch ungünstigere Anordnung mit m = -½ durch die Spinfunktion β wiedergegeben wird. Der Energieunterschied der beiden Energieniveaus beträgt:
ΔE = Eβ - Eα = γ · (h/2π) · B0
Diese Gleichung und die folgende schematische Darstellung zeigt, dass ΔE direkt von B0 abhängig ist.
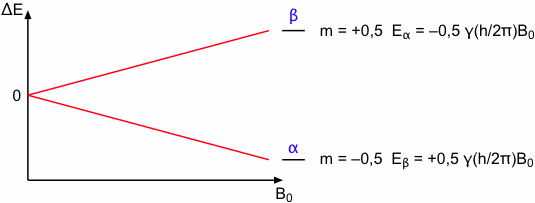
Mit der Existenz energetisch unterschiedlicher Zustände ist die Voraussetzung für die Spektroskopie gegeben. Durch Zufuhr eines entsprechenden Energiequants kann ein Übergang vom energieärmeren α in das energiereichere β Niveau erreicht werden. Die Resonanzbedingung lautet:
ΔE = h · ν = γ · (h/2π) · B0
Die Resonanzfrequenz für Protonen liegt bei einem Magnetfeld von 2,35 T bei 100 MHz, was einer Radiowelle mit einer Wellenlänge von 3 m entspricht. Andere NMR-aktive Kernsorten besitzen eigene charakteristische Resonanzfrequenzen. Da der Energieunterschied zwischen den Kern-Zeeman-Niveaus bei Raumtemperatur sehr klein ist, sind die beiden Niveaus nahezu gleich besetzt. Der Überschuss im energieärmeren Niveau liegt lediglich im Bereich von 10-6 % (ppm). Dies ist eine Ursache für die geringe Empfindlichkeit der NMR-Spektroskopie.
Nicht nur die Kernsorte, sondern auch die Molekülumgebung hat einen entscheidenden Einfluss auf die Resonanzfrequenz NMR-aktiver Kerne. So unterscheiden sich z.B. die Anregungsenergien von Protonen je nach deren Lage in einem Molekül auf charakteristische Weise. Darüber hinaus liefern neben der Lage der Peaks in einem NMR-Spektrum auch deren Feinstruktur (Multiplizität), Breite und Intensität wichtige Informationen. Aufgrund der hohe Aussagekraft von NMR-Spektren hat sich die NMR-Spektroskopie zu einem der wichtigsten Verfahren in der Strukturanalyse entwickelt.
Instrumentelles
In der NMR-Spektroskopie unterscheidet man zwei prinzipiell unterschiedliche Aufnahmetechniken. Bei der inzwischen veralteten CW-Technik (continuous wave) wird die in einem geeigneten deuterierten Lösungsmittel, z.B. CDCl3, gelöste Probe in einem rotierenden NMR-Röhrchen einem möglichst homogenen Magnetfeld ausgesetzt. Die Anregung der Probe geschieht durch einen Hochfrequenzsender. In der senkrecht zur Senderspule und zum Magnetfeld angeordneten Empfangsspule wird im Resonanzfall durch die in der Probe bei der Spininversion induzierte Änderung der Magnetisierung ein Strom erzeugt. Das entsprechende Signal wird verstärkt und registriert. Zur Erfüllung der Resonanzbedingung kann man entweder bei konstanter Feldstärke B0 die Frequenz ν variieren (Frequenz-Sweep) oder bei konstanter Frequenz ν0 das Magnetfeld B (Feld-Sweep). Der schematische Aufbau eines CW-NMR-Spektrometers ist in der Vorlage zu diesem Kapitel abgebildet.
Beim FT-Verfahren (Fourier-Transformation) werden durch eine Sequenz von Hochfeldpulsen alle Kerne einer Kernsorte, also z.B. alle Protonen, gleichzeitig angeregt. Dadurch wird die makroskopische Magnetisierung der Moleküle in Feldrichtung geändert. Nach dem Puls stellt sich die Gleichgewichtssituation wieder ein, womit eine zeitabhängige Änderung der Magnetisierung verbunden ist, welche registriert wird. Aus dem resultierenden Free-Induction-Decay (FID), einem komplexen Interferogramm aus überlagerten Schwingungen, kann man durch eine Fourier-Transformation ein normales Kernresonanz-Spektrum berechnen. Das Signal wird dabei von der Zeitdomäne in die Frequenzdomäne transformiert.
Die Zeit für einen Impuls und die Wartezeit für die Aufnahme des FID ist so kurz, dass eine große Anzahl von FIDs akkumuliert werden können, bevor die Fourier-Transformation durchgeführt wird. Daraus resultiert eine enorme Verbesserung der Empfindlichkeit und eine Verbesserung des Signal/Rausch-Verhältnisses. Darüber hinaus ist durch den Einsatz geeigneter Pulsfolgen die Aufnahme spezieller ein- und zweidimensionaler NMR-Spektren möglich, welche mit der CW-Technik nicht zugänglich sind.
1H-NMR-Spektroskopie
Das 1H-NMR Spektrum
Als Beispiel ist in der folgenden Abbildung das 1H-NMR Spektrum von 4-Methylbenzoesäureethylester abgebildet.
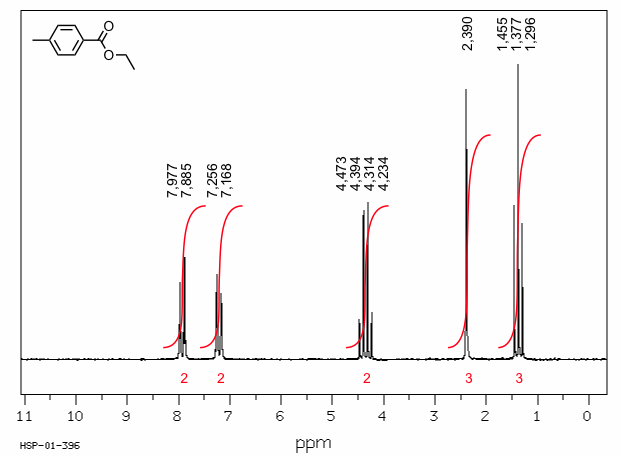
[Quelle: SDBS (Integrated Spectral Data Base System for Organic Compounds), National Institute of Advanced Industrial Science and Technology Japan]
Die Peaks in diesem Spektrum können den Protonen in dieser Verbindung folgendermaßen zugeordnet werden.
| δ | Multiplizität | J | Zuordnung |
| 7,93 | Dublett | 3J = 8,3 Hz | aromatische Protonen in 2-Position |
| 7,21 | Dublett | 3J = 8,2 Hz | aromatische Protonen in 3-Position |
| 4,35 | Quartett | 3J = 7,1 Hz | CH2-Gruppe der Ethylgruppe |
| 2,39 | Singulett | Methylgruppe am aromatischen Ring | |
| 1,38 | Triplett | 3J = 7,1 Hz | CH3-Gruppe der Ethylgruppe |
Ein 1H-NMR-Spektrum ist vollständig interpretiert, wenn man allen chemisch nicht äquivalenten Protonen der betreffenden Verbindung ein Signal mit Angabe der chemischen Verschiebung δ (oder bei breiten Signalen bzw. Multipletts des Bereichs der chemischen Verschiebung) zuordnet. Daneben sollte jedes Signal durch seine Multiplizität (Singulett, Dublett, Triplett usw.) und gegebenenfalls durch eine oder mehrere Kopplungskonstanten charakterisiert werden. Letztere werden in Hertz angegeben. Die chemische Verschiebung δ ist eine dimensionslose Größe, die folgendermaßen definiert ist:
δ = [ν(x) - ν(TMS)] / ν(TMS) · 106
ν(x) steht für die Resonanzfrequenz des Protons Hx und ν(TMS) für die der Standardreferenzsubstanz in der 1H-NMR-Spektroskopie, dem Tetramethylsilan [Si(CH3)4, TMS]. Näherungsweise kann man im Nenner der Definition von δ die Messfrequenz des verwendeten NMR-Spektrometers einsetzen. Mit dieser Vereinfachung erhält man:
δ = [ν(x) - ν(TMS)] / Messfrequenz · 106
Die chemische Verschiebung ist also von der Messfrequenz des Spektrometers unabhängig. Außerdem ist δ(TMS) = 0, da [ν(x) - ν(TMS)] = 0. Die δ-Werte in einem 1H-NMR-Spektrum werden ausgehend von dem TMS-Signal links positiv und rechts negativ angegeben. Verwendet man als Ordinate eines Spektrums die Frequenzskala, so nimmt die Frequenz von rechts nach links also zu. Die Feldstärkeskala ist genau gegenläufig. Die Verschiebung eines Signals "zu hohem Feld" ("Hochfeldverschiebung") bedeutet z.B. also eine Verschiebung des Signals im Spektrum nach rechts.
Chemische Verschiebung
Die exakte Resonanzfrequenz eines Protons hängt in charakteristischer Weise von der elektronischen Umgebung des betreffenden Kerns ab. Die Ursache hierfür ist das lokale Magnetfeld, welches durch die den Kern umgebenden Elektronen hervorgerufen wird und welches dem äußeren Magnetfeld entgegengerichtet ist. Die Konsequenz dieses Magnetfeldes ist eine geringere effektive Feldstärke Beff in der Umgebung des Kerns, wodurch sich für die Resonanzbedingung folgender korrigierter Ausdruck ergibt:
ν = (γ/2π) · Beff
Das Proton wird durch die Elektronen also abgeschirmt. Das Ausmaß der Abschirmung kann man durch eine dimensionslose Abschirmkonstante σ berücksichtigen.
Beff = B0 - σ · B0
ν = (γ/2π) · (1 - σ) · Beff
Je stärker ein Kern abgeschirmt wird, je größer also σ ist, desto kleiner wird Beff und desto größer muss bei konstanter Frequenz das angelegte Feld sein, um den Kern in Resonanz zu bringen. Eine analoge Überlegung zeigt, dass die Resonanzfrequenz ν bei konstantem Feld B0 mit wachsender Abschirmung abnimmt.
Alle Effekte, die die Elektronendichte an einem Kern beeinflussen, wirken sich also auf die chemische Verschiebung des betreffenden Kern aus. Im Einklang mit den obigen Betrachtungen findet man, dass elektronegative Substituenten, die die Elektronendichte an einem Kern herabsetzen, den Kern entschirmen, die chemische Verschiebung also vergrößern. Diese Effekte werden in dem von Shoolery entwickelten Inkrementsystem berücksichtigt, mit dem die chemische Verschiebung von Methylenprotonen in Verbindungen der allgemeinen Struktur X-CH2-Y abgeschätzt werden kann (siehe Vorlage).
Bei ungesättigten Systemen wie Alkenen, Carbonyl-Verbindungen, Alkinen oder Aromaten sind neben induktiven auch die mesomeren Effekte der Substituenten zu berücksichtigen. Daneben treten in solchen Systemen sogenannte Anisotropieeffekte auf, die Protonen, welche sich in bestimmten Bereichen um die π-Systeme der ungesättigten Verbindungen befinden, entschirmen oder abschirmen. Schematisch kann man diese Effekte, wie in der folgenden Abbildung gezeigt, symbolisieren. Dabei werden die Protonen, die sich in dem mit – gekennzeichneten Bereich befinden entschirmt und solche, die in dem mit + gekennzeichneten Bereich lokalisiert sind, abgeschirmt. Eine Erklärung der starken Entschirmung von Protonen in der Ringebene von Hückel-Aromaten liefert das Ringstrommodell.
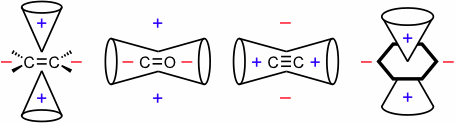
Zur Berechnung der chemischen Verschiebung von Protonen an olefinischen Doppelbindungen und an Benzolringen sind ebenfalls Inkrementsysteme entwickelt worden, die in der Vorlage zu diesem Kapitel angegeben sind.
Die Zahl der in einem 1H-NMR-Spektrum auftretenden Signale wird durch die Symmetrie und Flexibilität des Moleküls bestimmt. Zwei Kerne eines Moleküls sind chemisch äquivalent und besitzen dadurch dieselbe Resonanzfrequenz (sie sind "isochron"), wenn sie durch eine auf das Molekül anwendbare Symmetrieoperation ineinander überführt werden können oder wenn sie durch eine schnelle intramolekulare Bewegung, z.B. eine Rotation um eine Einfachbindung, im Zeitmittel identisch werden. Ein nützliches Verfahren, um die chemische Äquivalenz von Protonen in einer organischen Verbindung zu überprüfen, besteht in der Substitution jedes dieser Protonen durch eine Testgruppe. Führen dabei alle betreffenden Wasserstoffatome jeweils zur selben Verbindung (oder zu einem Enantiomerenpaar), sind die Wasserstoffatome chemisch äquivalent und haben dieselbe chemische Verschiebung.
Bei Methylenprotonen unterscheidet man homotope, enantiotope und diastereotope Protonen. Homotope Protonen geben bei Anwendung des Substitutionskriteriums dieselbe Verbindung, enantiotope Protonen ein Enantiomerenpaar und diastereotope Protonen ein Diastereomerenpaar. Homotope Protonen sind 1H-NMR-spektrokopisch nicht unterscheidbar, enantiotope Protonen in einer achiralen Umgebung ebenfalls nicht. Diastereotope Protonen sind jedoch chemisch nicht äquivalent und besitzen in der Regel unterschiedliche chemische Verschiebungen. Folgende Abbildung zeigt diesen Zusammenhang an einigen Beispielen. Beim letzten Molekül ist zu beachten, dass die Protonen der markierten Methylengruppe diastereotop sind, obwohl im Molekül kein weiteres stereogenes Zentrum vorhanden ist. Die Anwendung des Substitutionskriteriums zeigt allerdings, dass durch Ersatz des einen und des anderen Protons durch eine Testgruppe gleichzeitig zwei stereogene Zentren gebildet werden, da die Äquivalenz der beiden CH2OH-Gruppen aufgehoben wird.
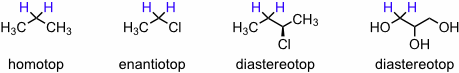
Die chemische Äquivalent von Protonen in cyclischen Verbindungen ergibt sich aus der Flexibilität dieser Systeme. So gehen bei Raumtemperatur die axialen und äquatorialen Protonen in der Sesselkonformation des Cyclohexan durch Ringinversion schnell ineinander über. Solange solche Vorgänge schnell auf der NMR-Zeitskala sind, beobachtet man im Spektrum nur ein einziges, scharfes, gemitteltes Signal. Durch Abkühlen kann man solche Prozesse aber oft so verlangsamen, dass sie NMR-spektroskopisch aufgelöst werden können. Die temperaturabhängige NMR-Spektroskopie kann auf diese Weise z.B. ausgenutzt werden, um die freie Aktivierungsenthalpie von konformationellen Umwandlungen zu bestimmen.
Intensität
Die Fläche unter einem NMR-Signal ist ein Maß für die Intensität eines Übergangs und damit in 1H-NMR-Spektren proportional zu der Zahl der an diesem Übergang beteiligten Protonen. Die Integrale von Peakflächen werden in 1H-NMR-Spektren in Form von Stufenkurven berechnet. Ein Vergleich der Höhe dieser Kurven für verschiedene Signale liefert unmittelbar Informationen über die relativen Verhältnisse der an den einzelnen Absorptionen beteiligten, chemisch äquivalenten Protonen.
Linienbreite
Die Peaks in 1H-NMR-Spektren sind meist schmal. Auffällige Peakverbreiterungen können apparative Ursachen haben (Feldinhomogenitäten), aber auch strukturelle. So kommt es zu einer Verbreiterung der NMR-Signale, wenn intramolekulare oder intermolekulare Austauschprozesse Geschwindigkeiten besitzen, die im Bereich der NMR-Zeitskala liegen und daher nicht mehr aufgelöst werden können. Zu intermolekularen Austauschprozessen zählt der Protonentransfer, bei intramolekularen Austauschprozessen kann es sich um konformationelle Umwandlungen oder Valenzisomerisierungen handeln.
Spin-Spin-Kopplung
Die in der Protonenresonanz gemessenen Signale zeigen häufig eine Feinstruktur. Nach der Anzahl der Teilbanden im Signal spricht man von einem Singulett, einem Dublett, einem Triplett, einem Quartett, einem Quintett usw. (bei vielen Linien Multiplett). Ursache für die Aufspaltung von Signalen ist die Wechselwirkung eines Kerns mit Nachbarkernen. Diese Spin-Spin-Kopplung tritt zwischen Kernen derselben Sorte auf (homonuklear) und zwischen Kernen verschiedener Elemente (heteronuklear). Sie wird durch die Beeinflussung der Resonanz eines Kerns A durch das lokale Magnetfeld des koppelnden Kerns X verursacht.
Vereinfacht kann man den Effekt folgendermaßen erklären: sind zwei Kerne A und X so weit voneinander entfernt, dass sie sich gegenseitig nicht beeinflussen, beobachtet man für beide Kerne Singuletts bei den charakteristischen Resonanzfrequenzen. Ist allerdings der Abstand klein genug und die beiden lokalen Magnetfelder von A und X beeinflussen einander, sind die Signale der beiden Kerne zu jeweils einem Dublett aufgespalten. Ursache hierfür ist, dass die Anregungsenergie beider Kerne vom Zustand des jeweilig anderen abhängt. Ist Kern X z.B. im α-Zustand, so führt das lokale Feld von X zur Verringerung der zur Anregung von A vom α- in den β-Zustand notwendigen Energie, da X das Feld von A verstärkt. Umgekehrt ist, wenn sich X im β-Zustand befindet, das Feld von X dem von A entgegengerichtet, so dass zur Resonanz von A ein höherer Energiebetrag notwendig ist und sich das entsprechende Signal zu höherem Feld verschiebt. Als Ergebnis erhält man also zwei Dubletts, in dem die beiden Linien jeweils denselben Abstand besitzen. Dieses einfache Spinsystem wird AX-System genannt. Es handelt sich um ein Spektrum erster Ordnung.
Das Ausmaß der Kopplung wird quantitativ durch Angabe der Kopplungskonstante J beschrieben, die in Hertz angegeben wird. Die Kopplungskonstante ergibt sich bei dem obigen AX-System aus den Abständen der beiden Linien in den Dubletts, welche jeweils gleich sein müssen. In Abhängigkeit der Anzahl der Bindungen zwischen zwei miteinander Koppelnden Kernen unterscheidet man 1J-Kopplungen (direkte (heteronukleare) Kopplungen), 2J-Kopplungen (geminale Kopplungen, 8 - 18 Hz) und 3J-Kopplungen (vicinale Kopplungen, 6 - 8 Hz). Kopplungen über mehr als drei Bindungen sind meist vernachlässigbar außer in speziellen Systemen, in denen solche sogenannten Fernkopplungen signifikante Kopplungskonstanten besitzen können. In der 1H-Spektroskopie sind vor allem 3J-Kopplungen von Bedeutung. Die Größe dieser Kopplungskonstanten hängt von dem Abstand der beteiligten Kerne, den Bindungswinkeln und dem Torsionswinkel ab. Einen quantitativen Zusammenhang zwischen Torsionswinkel und der 3J-Kopplungskonstante liefert die Karplus-Gleichung.
Spinsysteme, an denen m isochrone Kerne A und n chemisch und magnetisch äquivalente Kerne X beteiligt sind, bezeichnet man als AmXn-Systeme. Aufgrund der Zunahme der Einstellungsmöglichkeiten aller Kerne A und aller Kerne X zueinander, liefert solche Systeme höher aufgespaltene Signale. Dabei ergibt sich die Multiplizität des Signals von A zu n + 1 und des Signals von X zu m + 1. (Allgemein gilt Multiplizität = 2 · I · m + 1, wobei m die Anzahl der koppelnden Nachbarkerne ist. Da in der 1H-NMR-Spektroskopie I = ½, vereinfacht sich die Beziehung zu m + 1). Das Signal eines Kerne hat also immer eine Linie mehr als koppelnde isochrone Nachbarkerne. Für die Methylgruppe des Ethanols ergibt sich im 1H-NMR-Spektrum also ein Triplett und für die Methylengruppe ein Quartett. Die magnetische Äquivalenz von koppelnden Kernen ist nur gegeben, wenn jeder chemisch äquivalente Kern A zu jedem chemisch äquivalenten Kern X dieselbe Kopplungskonstante besitzt. Kopplungen zwischen magnetisch äquivalenten Kernen treten in einem NMR-Spektrum nicht in Erscheinung.
Neben der Linienanzahl ist für ein Signal mit Feinstruktur auch die Intensitätsverteilung der einzelnen Linien charakteristisch. In der 1H-NMR-Spektroskopie entsprechen die relativen Intensitäten von Multipletts den Binominalkoeffizienten, welche dem Pascalschen-Dreieck zu entnehmen sind.
| Anzahl der koppelnden Nachbarkerne | Anzahl der Linien | Relative Intensitäten |
| 0 | 1 (Singulett) | 1 |
| 1 | 2 (Dublett) | 1 : 1 |
| 2 | 3 (Triplett) | 1 : 2 : 1 |
| 3 | 4 (Quartett) | 1 : 3 : 3 : 1 |
| 4 | 5 (Quintett) | 1 : 4 : 6 : 4 : 1 |
| 5 | 6 (Sextett) | 1 : 5 : 10 : 10 : 5 : 1 |
Bei Spektren erster Ordnung ergibt sich die chemische Verschiebung eines Kerns aus dem Schwerpunkt (Mittelpunkt) des Signals und die Kopplungskonstante aus dem Abstand zweier Linien.
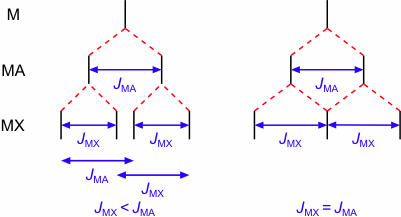
Bei Kopplung eines Protons M mit zwei chemisch nicht äquivalenten Protonen A und X erhält man (sofern Δν(MA)/JMA > 10 und Δν(MX)/JMX > 10) ein AMX-System. Darin wird das Signal von M durch die Kopplung mit A zu einem Dublett aufgespalten und die beiden Linien dieses Dubletts werden durch die Kopplung mit X erneut aufgespalten. Als Resultat erhält man ein Dublett vom Dublett, wobei die Kopplungskonstanten JMA und JMX wie in folgender Abbildung gezeigt bestimmt werden können. Ist zufällig JMA = JMX, dann beobachtet man für das Signal von M ein Multiplett, dessen Linienzahl sich gemäß der m + 1 Regel ergibt, wobei m die Summe aller dem Proton M benachbarten Protonen ist. In einem AMX-System mit JMA = JMX beobachtet man für das Proton M also ein Triplett.
Für die Kopplung von Kernen magnetisch nicht äquivalenter Kerne verlieren die einfachen Regeln, welche für Spektren erster Ordnung gelten, ihre Gültigkeit. Man erhält in solchen Fällen Spektren höherer Ordnung, die meist komplexe Aufspaltungsmuster besitzen. Ein Beispiel für eine Verbindung mit magnetisch nicht äquivalenten Protonen ist das para-Nitrophenol. Darin sind die beiden chemisch äquivalenten ortho-Protonen magnetisch nicht äquivalent, da das eine ortho-Proton zu einem meta-ständigen Proton über eine 3J-ortho-Kopplung koppelt, während es sich bei der Kopplung des anderen ortho-Protons mit demselben meta-Proton um eine 5J-para-Kopplung handelt. Analoges gilt für die Kopplung der meta-Protonen mit den ortho-Protonen. Magnetisch nicht äquivalente Protonen werden mit einem Strich gekennzeichnet. Das Spinsystem des para-Nitrophenols ist dementsprechend ein AA'XX'-System.
Ein weiteres Kriterium für das Auftreten von Spektren höherer Ordnung ist der Quotient aus dem Linienabstand der Signale zweier miteinander koppelnder Protonen und der Kopplungskonstante Δν/J. Ist Δν/J < 10, so erhält man ebenfalls Spektren höherer Ordnung. Das einfachste Spinsystem dieser Art, an dem zwei miteinander koppelnde Kerne beteiligt sind, ist das AB-System. Dieses wird z.B. für diastereotope Protonen einer Methylengruppe beobachtet, die über eine 2J-Kopplung miteinander koppeln, sofern keine weiteren Kopplungspartner vorhanden sind. AB-Systeme bestehen aus zwei symmetrischen Dubletts, die einen ausgeprägten Dacheffekt aufweisen. Die Kopplungskonstante ergibt sich aus dem Abstand der Linien in den Dubletts. Die chemische Verschiebung jedes Protons liegt jeweils zwischen den Linien des entsprechenden Dubletts, wobei sie um so stärker zum intensiveren Signal verschoben ist, je stärker sich das Intensitätsverhältnis der beiden Signale unterscheidet. Zur genauen Berechnung der chemischen Verschiebung existiert eine einfache Beziehung.
13C-NMR-Spektroskopie
Das 13C-NMR Spektrum
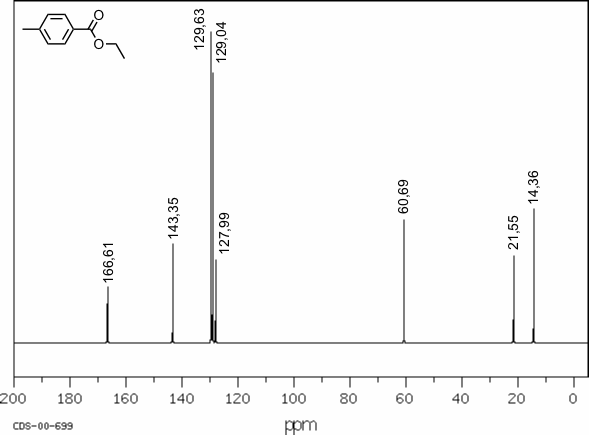
Als Beispiel ist in der folgenden Abbildung das 13C-NMR Spektrum von 4-Methylbenzoesäureethylester abgebildet.
[Quelle: SDBS (Integrated Spectral Data Base System for Organic Compounds), National Institute of Advanced Industrial Science and Technology Japan]
Die Peaks in diesem Spektrum können den C-Atomen in dieser Verbindung folgendermaßen zugeordnet werden. Dabei erfolgt die Zuordnung der C-Atome im aromatischen Ring durch Anwendung des entsprechenden Inkrementsystems (siehe Vorlage).
| δ | Zuordnung |
| 166,61 | Carbonyl C |
| 143,35 | C in Position 4 des aromatischen Rings |
| 129,63 | C in Position 2 des aromatischen Rings |
| 129,04 | C in Position 3 des aromatischen Rings |
| 127,99 | C in Position 1 des aromatischen Rings |
| 60,69 | C der CH2-Gruppe des Ethylesters |
| 21,55 | C der CH3-Gruppe am aromatischen Ring |
| 14,36 | C der CH3-Gruppe des Ethylesters |
Man erkennt, dass der Bereich der chemischen Verschiebung, den 13C-Absorptionen abdecken, erheblich größer ist als in 1H-NMR Spektren und üblicherweise von 0 bis 200 reicht. Ein weiterer wichtiger Unterschied ist, dass 13C-NMR-Spektren routinemäßig Breitband-entkoppelt aufgenommen werden, wodurch die 13C-1H-Kopplungen verschwinden und alle Signale im Spektrum als Singulett registriert werden. Da aufgrund dieser Entkopplung die einzige Information, die man einem Signal in einem 13C-Spektrum entnehmen kann, die chemische Verschiebung ist, ist es bei dieser Art der NMR-Spektroskopie besonders wichtig, den Zusammenhang zwischen chemischer Verschiebung und der Struktur eines Moleküls zu kennen. Als Referenzsubstanz zur Fixierung der δ-Skala wird wie in der 1H-Spektroskopie Tetramethylsilan (TMS) verwendet. Man definiert δ(13C von TMS) = 0.
Chemische Verschiebung
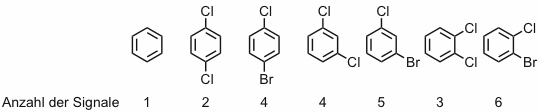
Wie in der 1H-NMR-Spektroskopie besitzen auch in der 13C-NMR-Spektroskopie chemisch äquivalente Kerne dieselbe chemische Verschiebung. Dies gilt sowohl für starre Moleküle wie für flexible, in denen z.B. einzelne Gruppen aufgrund von schneller Rotation um Einfachbindungen ununterscheidbar werden. Ein Beispiel für den zweiten Fall sind die Methylgruppen in einer tert-Butylgruppe. Die folgende Abbildung zeigt, dass durch Molekülsymmetrie die Anzahl der Signale in einem 13C-NMR-Spektrum geringer als die Anzahl der C-Atome in dem betreffenden Molekül sein kann.
Die wichtigsten Parameter, die die Absorptionsfrequenz von 13C-Kernen beeinflussen sind Hybridisierung und Ladungsdichte. Die Abhängigkeit der 13C-Resonanzfrequenz von der Hybridisierung entspricht der von gesättigten, acetylenischen und olefinischen Protonen in der 1H-NMR-Spektroskopie, d.h. sp3-C-Atome absorbieren bei höchstem Feld, dann folgen sp- und sp2-C-Atome. Ein besonderer Einfluss des Ringstroms in aromatischen Systemen auf die Resonanz der 13C-Atome in der Ringebene wird im Gegensatz zur 1H-NMR-Spektroskopie nicht beobachtet; olefinische und aromatische 13C-Atome absorbieren im selben Bereich. Wie in der 1H-NMR-Spektroskopie gilt, dass Substituenteneffekte, die die Ladungsdichte an einem 13C-Kerne herabsetzen, diesen entschirmen, seine chemische Verschiebung also erhöhen. Dabei muss man zwischen induktiven und mesomeren Effekten unterscheiden. Induktive Effekte haben nicht nur einen z.T. signifikanten Einfluss auf die chemische Verschiebung des direkt benachbarten 13C-Kernes, sondern auch auf weiter entfernte Kerne. Die konkreten Effekte einzelner Substituenten sind nicht immer leicht vorherzusagen, einen Anhaltspunkt gibt aber meist die Elektronegativität. Darüber hinaus existieren Inkrementsysteme, mit denen 13C-Resonanzen abgeschätzt werden können. Als Beispiel für ein solches Inkrementsystem wird in der Vorlage zu diesem Kapitel das für aromatische Systeme entwickelte vorgestellt. Eine steigende Alkylsubstitution führt im allgemeinen zu einer Tieffeldverschiebung der betreffenden 13C-Resonanz [δ(CH4) < δ(Cprim.) < δ(Csek.) < δ(Ctert.) < δ(Cquart.)]. Mesomere Effekte haben in der Regel einen Einfluss auf die Ladungsdichte spezifischer Positionen in einem Molekül und können diese Positionen entschirmen oder abschirmen, je nachdem ob sie die Ladungsdichte dort erniedrigen oder erhöhen.
Intensität
Routinemäßig werden in der 13C-NMR-Spektroskopie Messmethoden verwendet, die zu einer Verfälschung der Peakflächen der einzelnen Signale führen. Darum kann man aus der Integration der Signale nicht auf das relative Verhältnis der an den einzelnen Resonanzen beteiligten 13C-Kerne schließen. Hauptursache ist die 1H-Breitband-Entkopplung und die damit verbundenen Kern-Overhauser-Effekte.
Spin-Spin-Kopplung
Durch die 1H-Breitband-Entkopplung werden in 13C-NMR-Routinespektren die heteronuklearen 1H,13C-Kopplungen unterdrückt und alle Signale als Singulett reduziert. Durch diese 1H-Breitband-Entkopplung vereinfachen sich die 13C-NMR-Spektren erheblich, vorausgesetzt im Molekül sind keine anderen koppelnden Kerne wie Fluor, Phosphor oder Deuterium vorhanden. Die durch die 1H-Breitband-Entkopplung erzielten Vorteile (Zunahme der Signalintensität, Spektrenvereinfachung) wiegen den Informationsverlust, der durch den Wegfall der 13C,1H-Kopplung resultiert, auf, vor allem da heute zweidimensionale NMR-Methoden zur Verfügung stehen, z.B. H,C-COSY, durch die heteronukleare Kopplungspartner leicht zugeordnet werden können. Homonukleare 13C,13C-Kopplungen sind aufgrund der geringen Häufigkeit des 13C-Isotops (1,1 %) in Routinespektren nicht sichtbar.
Materialien
| zu Kapitel 3.1. | NMR-aktive Kerne |
| zu Kapitel 3.2. | Magnetische Resonanz Tomographie |
| zu Kapitel 3.3. | NMR-Spektrometer |
| zu Kapitel 3.4.1. | 1H-NMR-Spektrum |
| zu Kapitel 3.4.2. | Ringstrommodell |
| zu Kapitel 3.4.2. | Anisotropieeffekte |
| zu Kapitel 3.4.2. | Inkrementsysteme für 1H-Resonanz |
| zu Kapitel 3.4.5. | Karplus Kurve |
| zu Kapitel 3.5.1. | 13C-NMR-Spektrum |
| zu Kapitel 3.5.2. | Inkrementsysteme für 13C-Resonanz |
Übungen
Spektreninterpretation
1H-NMR Spektroskopie
| 1. Übung | 1. Lösung |
| 2. Übung | 2. Lösung |
| 3. Übung | 3. Lösung |
13C-NMR Spektroskopie
| 1. Übung | 1. Lösung |
| 2. Übung | 2. Lösung |
| 3. Übung | 3. Lösung |
Aufgaben
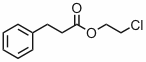
- Das 1H-NMR-Spektrum folgender Verbindung enthält neben den Signalen der aromatischen Protonen vier Tripletts. Welche beiden Möglichkeiten haben Sie (ohne Berücksichtigung von Inkrement-Systemen und Dacheffekten) eindeutig zu erkennen, welche beiden Paare von Tripletts jeweils den -CH2-CH2- Gruppen zuzuordnen sind?
Antwort - In der folgenden Tabelle sind die Frequenzen und chemischen Verschiebungen angegeben, die man im 1H-NMR Spektrum für die drei olefinischen Protonen HA, HB und HC des Styrols beobachtet. Berechnen Sie mit Hilfe dieser Informationen die chemischen Verschiebungen der drei Protonen und alle Kopplungskonstanten und ordnen Sie die Signale HA, HB und HC zu.
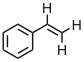
Hz ppm Hz ppm Hz ppm 615,63 6,874 521,56 5,824 473,44 5,287 604,88 6,754 520,50 5,812 472,31 5,274 598,06 6,678 504,00 5,628 462,69 5,167 587,31 6,558 502,88 5,616 461,46 5,154
Antwort - Bezeichnen Sie die Spinsysteme folgender Verbindungen nach der gängigen Nomenklatur.
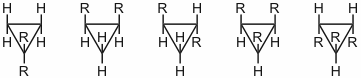
Antwort - Bezeichnen Sie die Spinsysteme folgender Verbindungen nach der gängigen Nomenklatur.
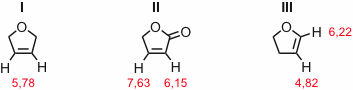
Antwort - Erklären Sie, auf welche Effekte die chemischen Verschiebungen der olefinischen Protonen in II und III im Vergleich zu I zurückzuführen sind.
Antwort - In Breitband-entkoppelten 13C-NMR Spektren ist das Signal des C-Atoms von CDCl3 zu einem Triplett aufgespalten. Warum? Welche Multiplizität hat das Signal der C-Atome in d6-DMSO [(D3C)2S=O]?
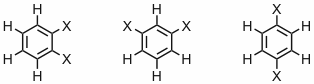
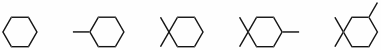
Antwort - Wie viele Signale erwarten Sie in den 13C-NMR Spektren folgender Verbindungen?
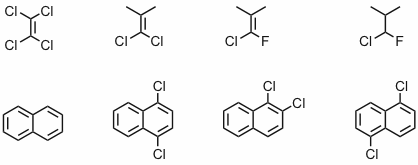
Antwort - Wie viele Signale erwarten Sie in den 13C-NMR Spektren folgender Verbindungen?
Antwort - Die C-Atome der beiden markierten Methylgruppen in Verbindung I sind im 13C-NMR Spektrum chemisch äquivalent, die in Verbindung II nicht. Erklären Sie warum.
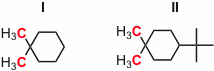
Antwort
Literatur
- S. Berger, S. Braun, 200 and More NMR Experiments - A Practical Course, Wiley-VCH, Weinheim, 2004
- E. Breitmaier, Vom NMR-Spektrum zur Strukturformel organischer Verbindungen, Wiley-VCH, Weinheim, 2005
- H. Friebolin, Ein- und zweidimensionale NMR Spektroskopie, VCH, Weinheim, 1988
- H. Günther, NMR-Spektroskopie, 3. Auflage, Thieme Verlag, Stuttgart, 1992
- M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in der Organischen Chemie, 6. Auflage, Thieme Verlag, Stuttgart, 2002
- J. B. Lambert, E. P. Mazzola, Nuclear Magnetic Resonance Spectroscopy - An Introduction to Principles, Applications, and Experimental Methods, Pearson Prentice Hall, Upper Saddle River, NJ, 2004
- T. N. Mitchell, B. Costisella, NMR - From Spectra to Structures, Springer Verlag, Berlin, 2004
- D. H. Williams, I. Fleming, K.-P. Zeller, Strukturaufklärung in der organischen Chemie, 6. Überarbeitete Auflage, Thieme Verlag, Stuttgart, 1991
Online-Datenbanken mit einer Vielzahl von 1H- und 13C-NMR Spektren organischer Verbindungen:
Weitere Informationen und Online-Kurse zur NMR-Spektroskopie finden sich unter folgenden Links:
- InternetChemie
- Vernetztes Studium - Chemie
- Anwendung der 1H-NMR-Spektroskopie (TU München)
- NMR Lectures Pages (University of the Sciences in Philadelphia)
- NMR Course (Queen's University, Ontario, Canada)
- NMR Spectroscopy (Rochester Institute of Technology)
- NMR Spectroscopy - Principles and Application (Imperial College London)
Letzte Änderung: 23-03-30. Email