Lehre - Supramolekulare Chemie - Wirt-Gast-Systeme
Coronanden
Als Coronanden (oder Kronenether) werden cyclische Polyether aus mit Ethylenbrücken verknüpften Sauerstoffatomen bezeichnet.
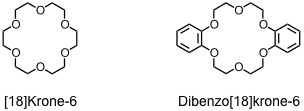
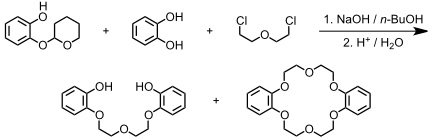
Kronenether waren eine Zufallsentdeckung. Sie wurden von C. J. Pedersen als Nebenprodukte bei folgender Veretherung beobachtet, bei der als Verunreinigung im Reaktionsgemisch auch ungeschütztes Brenzcatechin vorhanden war.
Pedersen stellte fest, dass Natrium- oder Kaliumsalze in unpolaren Lösungsmitteln wie Benzol oder Chloroform nach Zusatz eines Kronenethers gelöst werden können. Er erklärte dies dadurch, dass der Kronenether das Kation des Salzes komplexiert. Aus Elektroneutralitätsgründen folgt das Anion dem solvatisierten Kation dann in die organische Phase.
Fast gleichzeitig mit der Entdeckung der Kronenether wurden erstmals makrocyclische Naturstoffe (Ionophore) identifiziert, die Alkalimetallkation in der unpolaren Umgebung biologischer Membranen solvatisieren können. Beispiele sind Nonactin und Valinomycin.
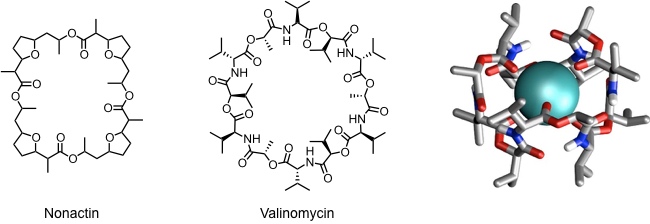
[Für eine interaktive Version der Kristallstruktur hier klicken]
[Quelle: V. Z. Pletnev, I. N. Tsygannik, Y. D. Fonarev, I. Y. Mikhailova, Y. V. Kulikov, V. T. Ivanov, D. A. Langs, W. L. Duax Bioorg. Khim. 1995, 21, 828-833.]
Ähnlich wie beim Valinomycin beruht die Bindung von Kronenethern an ihre Gäste auf Ion-Dipol-Wechselwirkungen zwischen den Sauerstoffatomen entlang des Rings und dem in den Hohlraum eingelagerten Kation.
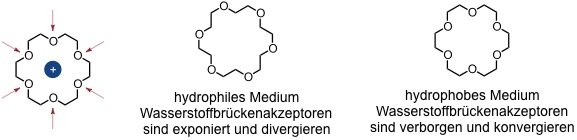
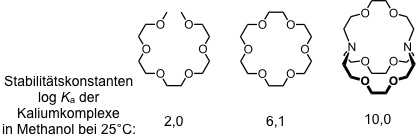
In Abhängigkeit des Lösungsmittels bevorzugen Kronenether unterschiedliche Konformationen. Dieses Verhalten erklärt unter anderem, warum die Kationenaffinität von Kronenethern in unpolaren Lösungsmitteln erheblich höher ist als die in polaren. Die Stabilitätskonstante des Kaliumkomplexes von Dibenzo[18]krone-6 beträgt in Wasser z.B. 102 M-1, während sie in Methanol 106 M-1 erreicht.
Die Kationenselektivität von Kronenethern wird im Wesentlichen von der Ringgröße bestimmt.
| Kation | Ionendurchmesser / pm | Kronenether | Hohlraumdurchmesser / pm |
| Li+ | 120 | [12]Krone-4 | 120-150 |
| Na+ | 190 | [15]Krone-5 | 170-220 |
| K+ | 266 | [18]Krone-6 | 260-320 |
| Cs+ | 338 | [21]Krone-7 | 340-430 |
Komplexstabilität in Abhängigkeit der Ringgröße in Methanol
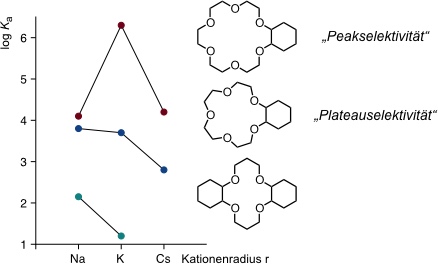
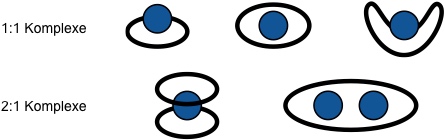
Je nach Verhältnis von Ringgröße des Kronenethers und Durchmesser des Kations werden verschiedene Komplexstrukturen und Komplexstöchiometrien beobachtet.
Enantioselektive Erkennung
Die enantioselektive Komplexierung chiraler Ammoniumionen gelingt mit chiralen Kronenetherderivaten. Der erste chirale Kronenether wurde in der Gruppe von D. J. Cram entwickelt. Er enthält als chirale Einheiten Binaphtholgruppen.
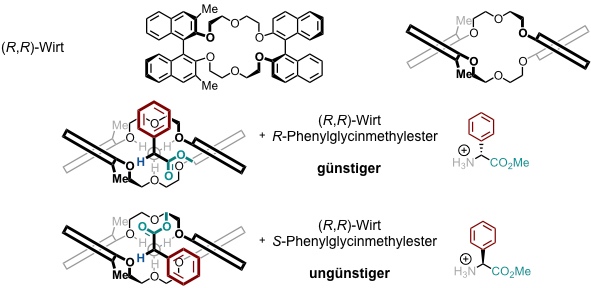
Das α-Wasserstoffatom des Substrats zeigt in Richtung der methylierten Binaphtholgruppe. Dann ist es günstiger, wenn die große Phenylgruppe in die größere Spalte zwischen den beiden Naphtholgruppen eingelagert wird.
Mit Hilfe dieses chiralen Kronenethers lassen sich die Racemate von Aminosäuren auch in präparativem Maßstab entweder durch Extraktion oder an einer stationären Phase chromatographisch trennen.
Die strukturelle Variationsbreite von Kronenethern ist sehr groß. So können:
- die Ringgröße verändert werden,
- Substituenten eingeführt werden,
- und die Donorstellen verändert werden. Neben Sauerstoff ist vor allem Stickstoff möglich (Azakronen), aber auch Schwefel, Phosphor und Arsen spielen eine gewisse Rolle. Auch die Heteroatome in Bauelementen wie Furan, Thiophen oder Pyridin können Donorfunktionen übernehmen.
Auf diese Weise können die Bindungseigenschaften von Kronenethern gezielt beeinflusst werden. So bevorzugen Kronenether mit Sauerstoffatomen als Donorstellen die härteren Kationen von Hauptgruppenelementen als Gäste, während Kronenether mit Schwefelatomen als Donorstellen zur Komplexierung von weicheren Übergangsmetallen, z.B. Ag+, besonders geeignet sind.
Kronenether oder Cryptanden können neben Metallkationen auch Ammoniumionen binden. Die Wechselwirkung ist dann besonders günstig, wenn die Anzahl komplementärer Bindungsstellen maximal ist.
Literatur
Cryptanden
Als Cryptanden bezeichnet man bicyclische oder polycyclische Kronenether. Diese Verbindungen wurden in den 70er Jahren des 20. Jahrhunderts maßgeblich von der Gruppe um J.-M. Lehn entwickelt.
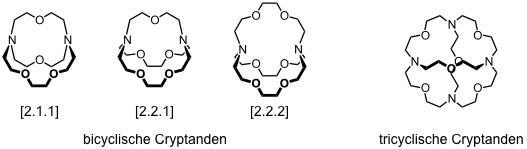
Bei Einlagerung von Kationen in den Hohlraum von Cryptanden wird der Gast von drei oder mehr Polyetherbrücken abgeschirmt. Als Folge dieser Verkapselung bilden Cryptanden generell stabilere Komplexe als Coronanden. Auch bei diesen Wirten sind Donorstellen- und Substituentenvariationen möglich.
Geeignete Kronenetherderivate können auch zur Komplexierung von Anionen dienen. Hierzu bedarf es jedoch Rezeptoren mit relativ großem Hohlraum, da Anionen einen größeren Durchmesser besitzen als Kationen (K
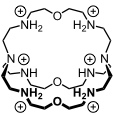
Für die Selektivität dieser Wirte ist wiederum die Komplementarität zwischen der Größe des Anions und der des Hohlraums mitverantwortlich.
Präparative Anwendungen von Coronanden und Cryptanden
Kronenether können Salze in organischen Lösungsmitteln solubilisieren. Da nur das Kation komplexiert wird, liegt das Anion in einem reaktiven Zustand vor (es ist „nackt“) und kann dadurch als Reagenz für eine Reihe von Reaktionen dienen. Dies gilt besonders bei Cryptaten, bei denen auch der Kontakt mit dem komplexierten Kation, welcher bei Kronenether- oder Podandkomplexen im Prinzip noch möglich ist, entfällt.
Solche Wirte können also Reaktionspartner, die sich aufgrund von unterschiedlicher Polarität nicht in einem gemeinsamen Lösungsmittel vereinigen lassen, in homogener Lösung zur Reaktion bringen. Dieses Prinzip ähnelt der Phasentransfer-Katalyse. Auf diese Weise verlaufen viele Reaktionen, die in Abwesenheit von Kronenethern nicht oder nur in geringen Ausbeuten durchführbar sind, nach deren Zusatz sehr effizient.
Jede Reaktion, bei der Ionen oder ionische Intermediate beteiligt sind, kann im Prinzip durch Kronenether beeinflusst werden. Beispiele sind:
- Nucleophile Substitutionen
- Reaktionen mit Carbanionen
- C–C Bindungsknüpfungen (z.B. Knoevenagel-Reaktionen)
- Additionen, Eliminierungen
- Carbenerzeugung (CHCl3/NaOH)
- Oxidationen, Reduktionen
- Umlagerungen, Isomerisierungen
- Polymerisationen
Bücher
- Gokel, G. W. "Crown Ethers and Cryptands" RSC: Cambridge, 1994.
Literatur
Podanden
Als Podanden bezeichnet man offenkettige Verbindungen, die in einer linearen oder verzweigten Kette Donoratome tragen.
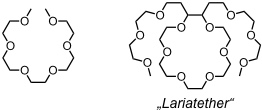
Podanden besitzen keinen für die Kationenbindung vororganisierten Hohlraum, sie können aber bei Komplexbildung eine Konformation mit einem Hohlraum annehmen. Da dies jedoch mit einem ungünstigen Entropiebeitrag verbunden ist, sind Komplexe von Podanden um Größenordnungen instabiler als die von Coronanden und diese wiederum stehen den Cryptandkomplexen und mehrere Zehnerpotenzen nach.
| Komplex | Ka / M-1 | Effekt |
| Podat | 102 - 104 | Chelateffekt |
| Coronat | 104 - 106 | makrocyclischer Effekt |
| Cryptat | 106 - 1010 | makrobicyclischer Effekt |
Unterschiede ergeben sich auch in der Kinetik ihrer Komplexbildung. Die konformativ starreren Cryptanden komplexieren langsamer als Coronanden und diese wiederum langsamer als die flexiblen Podanden.
Vororganisation
Rezeptoren, die auch in Abwesenheit des Gastes eine bereits für die Komplexbildung günstige Konformation besitzen, sind vororganisiert.
Der Entropieverlust bei der Komplexbildung (Verlust konformativer Freiheitsgrade) ist gering.
Dadurch ist bei analoger enthalpischer Stabilisierung eine höhere Gesamtstabilität für einen Komplex zu erwarten, als für den entsprechenden Komplex eines weniger gut vororganisierten Rezeptors.
Spheranden
Diese ebenfalls von D. J. Cram entwickelte Ligandenfamilie beruht auf einer cyclischen Anordnung von m- Phenyleneinheiten. Die Sauerstoffatome stellen wiederum Bindungsstellen für Kationen dar.
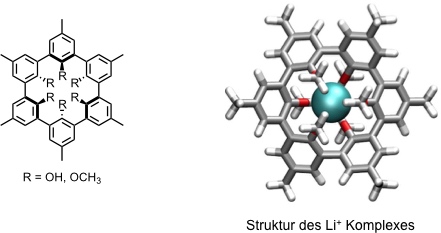
[Für eine interaktive Version der Kristallstruktur hier klicken]
Durch die starre Präorganisation der funktionellen Gruppen entsteht ein annähernd kreisförmiger Hohlraum. Dabei sind die sechs konvergent angeordneten funktionellen Gruppen alternierend up-down angeordnet. Der Durchmesser des Hohlraums variiert mit dem Diederwinkel der sechs Arengruppen. Im Mittel beträgt er 162 pm. Daher können Li+ (120 pm) und Na+ (190 pm) eingelagert werden, K+ (266 pm) jedoch nicht. Die Selektivität der Komplexbildung ist dadurch außerordentlich hoch.
Weitere von Cram hergestellte Spheranden unterscheiden sich in der Art der Heteroatome, in der Ringgröße, der Bindungsstellen oder der Konnektivität auf.
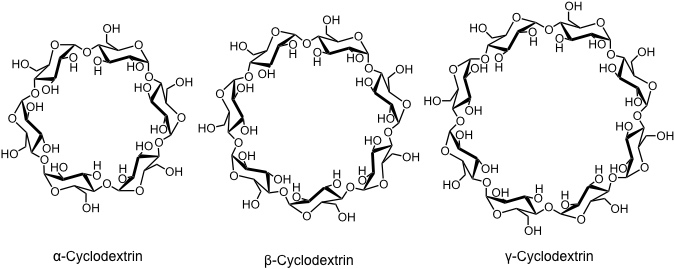
Cyclodextrine
Struktur
Cyclodextrine sind cyclische Oligosaccharide aus sechs (α), sieben (β) oder acht (γ) α-1→4 verknüpften D- Glucoseeinheiten. Cycodextrine mit neun oder mehr Anhydroglucoseeinheiten sind ebenfalls bekannt, sie spielen aber nur eine untergeordnete Rolle.
Cyclodextrine entstehen beim enzymatischen Abbau von Stärke durch spezielle Mikroorganismen. Diese Synthese dient auch zur industriellen Cyclodextrinherstellung, wobei für jedes Cyclodextrin der entsprechende Mikroorganismus und die Reaktionsbedingungen optimiert worden sind. Die Cyclodextrine fallen als Gemische mit erhöhtem Gehalt an dem gewünschten Cyclodextrin an und müssen im weiteren Verlauf der Gewinnung getrennt und gereinigt werden.
Cyclodextrine haben eine konische Molekülform mit einem definierten Hohlraum. Die primären OH-Gruppen sind am schmaleren Rand des Hohlraums und die sekundären am weiteren Rand angeordnet. In den Hohlraum weisen nur C–C und C–H Bindungen, wodurch das Innere des Hohlraums hydrophob ist.
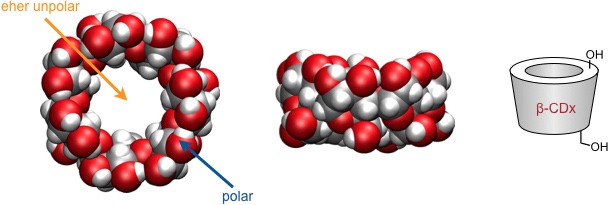
[Für eine interaktive Version der Kristallstruktur hier klicken]
Cyclodextrine sind wasserlöslich. In wässriger Lösung und im kristallinen Zustand sind sie in der Lage, unpolare Gastmoleküle in das Innere ihres Hohlraums aufzunehmen. Die Selektivität der Gastkomplexierung wird dabei im Wesentlichen durch die Größe des Hohlraums geprägt.
Die Konformation von β-Cyclodextrin wird durch intramolekulare Wasserstoffbrücken zwischen der 2-OH-Gruppe einer Anhydroglucoseeinheit und einer 3-OH-Gruppe der nächsten Anhydroglucoseeinheit stabilisiert.
Entdeckung
| 1891 | A. Villier. Erhielt eine neue Verbindung bei mikrobiellem Abbau von Stärke, für die er Summenformel (C6H10O5)2 · 3 H2O bestimmte und die er "Cellulosine" nannte. Er erkannte, dass es sich um einen nicht- reduzierenden Zucker mit besonderer Stabilität gegenüber saurer Hydrolyse handelt. |
| 1903 | F. Schardinger. Abbau von Stärke durch Bacillus macerans. Beobachtete die Färbung von Cyclodextrinen mit Iod, die eine Unterscheidung der verschiedenen Fraktionen erlaubt. |
| 1911 | H. Pringsheim. Berichtete erstmals über die Komplexierung organischer Verbindungen durch Cyclodextrine. |
| 1936 | K. Freudenberg. Arbeiten zur Strukturaufklärung. Postulierte die cyclische Struktur der Cyclodextrine. |
| ab 1950 | D. French, F. Cramer. Systematische Arbeiten zu Strukturen, Eigenschaften und Anwendungen von Cyclodextrinen. Danach exponentielle Zunahme von Arbeiten über Cyclodextrine. |
| ab 1970 | Cyclodextrine werden in größeren Mengen industriell hergestellt und finden auch aufgrund ihrer Unbedenklichkeit in Bezug auf Toxizität Anwendungen in unterschiedlichen Bereichen. |
Herstellung
Enzymatische Transformation von Maisstärke
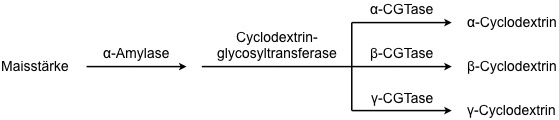
Weltproduktion aller Cyclodextrine ca. 5 × 103 T/Jahr
Preis
| Bulk-Preis in USD / kg | |
| α-Cyclodextrin | 45 |
| β-Cyclodextrin | 5 |
| γ-Cyclodextrin | 80 |
Bindungseigenschaften
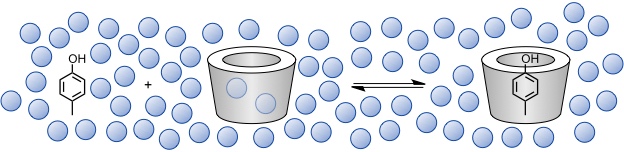
Die Stabilität von Cyclodextrinkomplexen beruht auf dem Zusammenwirken mehrerer Wechselwirkungsarten und hängt von der Substratstruktur und vom Lösungsmittel ab.
- Sterischer Fit
- Elektrostatische Wechselwirkungen
- Dipol-Dipol Wechselwirkungen
- van-der-Waals Wechselwirkungen
- Wasserstoffbrücken
Stabilität von β-Cyclodextrinkomplexen in Wasser (T = 298 K)
| Gast | log Ka |
| 1-Butanol | 1.2 |
| 1-Hexanol | 2.3 |
| Cyclohexanol | 2.8 |
| Benzol | 2.2 |
| Phenol | 3.4 |
| Benzoat | 1.1 |
| Adamantan-1-carboxylat | 4.6 |
[Quelle: Rekharsky, M. V.; Inoue, Y. Chem. Rev. 1998, 98, 1875-1917: <doi>.]
| Cyclodextrin | Anzahl Untereinheiten | Durchmesser des Hohlraums / pm | Durchmesser am weiteren Rand / pm | optimale Gäste |
| α | 6 | 570 | 1370 | Alkane, Benzol, Phenol |
| β | 7 | 780 | 1530 | 4-tert-Butylphenol, Naphthalin, Ferrocen, Adamantan |
| γ | 8 | 950 | 1690 | Perylen, Cholesterol, Kronenether |
Durch die Komplexbildung verändert sich die Löslichkeit der Cyclodextrine. So sind in der Regel nur die Komplexe von Gastmolekülen mit polaren Gruppen gut wasserlöslich. Je unpolarer das Gastmolekül ist, desto geringer ist die Löslichkeit des Komplexes. Bei Zugabe von p-Xylol zu einer wässrigen β-Cyclodextrinlösung fällt z.B. ein schwerlöslicher Komplex aus und da p-Xylol nur mit β-Cyclodextrin einen stabilen Komplex bildet, kann man diese Eigenschaft zur Abtrennung von β-Cyclodextrin aus einem Cyclodextringemisch ausnutzen.
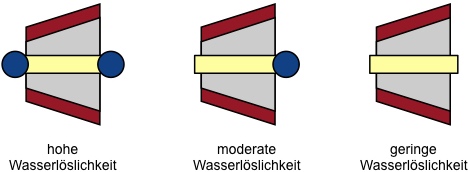
Derivatisierung
Cyclodextrine besitzen über die reine Größenerkennung von Gastmolekülen hinaus keine ausgeprägte Bindungsselektivität. Die Selektivität kann aber durch Einführung von Substituenten (bzw. Bindungsstellen) um den Cyclodextrinhohlraum gezielt verändert werden.
Statistische Substitution

Monosubstitution einer primären OH-Gruppe
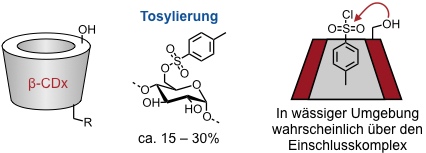
Ausgehend vom Tosylat können eine Vielzahl weiterer Derivate hergestellt werden: R = Hal, SH, N3, NH2, NR2, CHO, COOH usw.
Monosubstitution einer sekundären OH-Gruppe
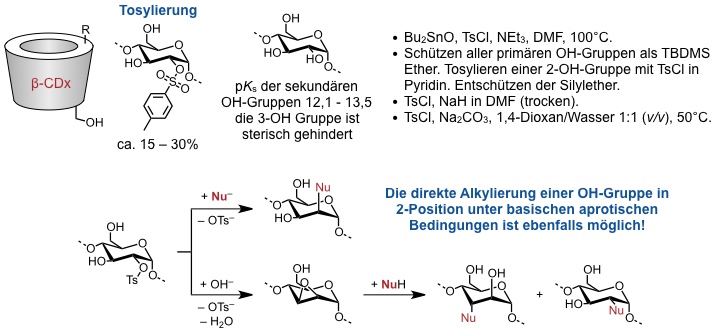
Mehrfachsubstitutionen
Die gezielte Tosylierung zweier Anhydroglucoseeinheiten im Ring gelingt durch Verwendung geeigneter Bis(sulfonsäurechloride). Dabei kann man durch die Struktur dieser Säurechloride steuern, ob benachbarte oder weiter entfernte Anhydroglucoseeinheiten modifiziert werden.
[Quelle: Khan, A. R.; Forgo, P.; Stine, K. J.; D’Souza, V. T. Chem. Rev. 1998, 98, 1977-1996: <doi>.]
Bis(cyclodextrine)
Durch kovalente Verknüpfung zweier Cyclodextrinringe kann die Komplexierung von Substraten mit langgestreckter Molekülgestalt deutlich verbessert werden.
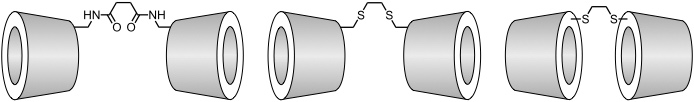
Dabei können die Bindungseigenschaften dieser Cyclodextrindimere durch die Spacerlänge und die gegenseitige Orientierung der Hohlraumöffnungen kontrolliert werden. Auch Heterodimere aus einer α- und einer β- Cyclodextrinuntereinheit sind beschrieben worden. In die Spacereinheiten solcher Cyclodextrindimere können außerdem funktionelle Gruppen eingebaut werden, die die chemische Modifizierung eines eingelagerten Gastmoleküls katalysieren.
[Quellen: Breslow, R.; Greenspoon, N.; Guo, T.; Zarzycki, R. J. Am. Chem. Soc. 1989, 111, 8296-8297: <doi>. Jiang, T.; Lawrence, D. S. J. Am. Chem. Soc. 1995, 117, 1857-1858: <doi>. Venema, F.; Nelissen, H. F. M.; Berthault, P.; Birlirakis, N.; Rowan, A. E.; Feiters, M. C.; Nolte, R. J. M. Chem. Eur. J. 1998, 4, 2237-2250: <doi>.]
Optische Sonden
Eine optische Detektion der Gastbindung gelingt bei Cyclodextrinen mit fluoreszenzaktiven Substituenten, z.B.Dansylgruppen.
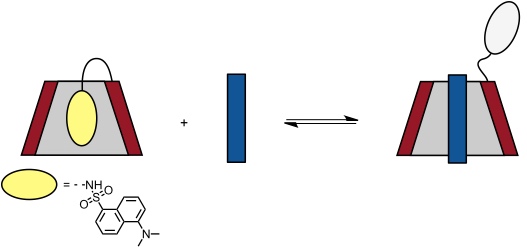
Der im Cyclodextrinhohlraum komplexierte Chromophor hat eine höhere Fluoreszenzintensität als außerhalb des Hohlraums, da das Innere des Hohlraums unpolar ist. Durch Einlagerung eines Gastes in den Hohlraum, welcher mit dem Cyclodextrin einen stabileren Komplex bilden muss als der Farbstoff, wird der fluoreszenzaktive Substituent verdrängt. Die damit verbundene direkt mit dem Auge detektierbare Abnahme der Fluoreszenzintensität der Lösung zeigt die Komplexbildung an.
Industrielle Anwendungen
Lebensmittelindustrie
- Schutz reaktiver Substanzen (Oxidation, Photooxidation, thermische Zersetzung)
- Eliminierung oder Reduktion von unangenehmem Geschmack oder Geruch
- Entfernung unerwünschter Komponenten aus Stoffgemischen (Cholesterol)
- Verbesserung der Löslichkeit schwer löslicher Komponenten
- Verlängerung der Haltbarkeit und Lagerfähigkeit
- Vereinfachung der Dosierung und Handhabung
Pharmaindustrie / Kosmetikindustrie
- Erhöhung der Bioverfügbarkeit durch Verbesserung der Wasserlöslichkeit
- Verbesserung der Dosierbarkeit
- Erhöhung der Stabilität
- ...
Textilindustrie
- Maskierung unangenehmer Gerüche auf Textilien durch Anbindung auf Textilfasern
- ...
Analytische Anwendungen
- Enantiomerentrennung auf Cyclodextrinphasen (HPLC / GC)
[Quelle: Hedges, A. R. Chem. Rev. 1998, 98, 2035-2044: <doi>.]
Literatur
Cyclophane
Struktur
Unter Cyclophanen versteht man Makrocyclen mit mehreren aromatischen Untereinheiten. Sie besitzen meist einen großen hydrophoben Hohlraum, in den neutrale oder positiv geladene Gastmoleküle eingelagert werden können. Ihre Bindungseigenschaften und ihr Löslichkeitsverhalten kann durch Einführung geeigneter Substituenten in weiten Grenzen variiert werden.
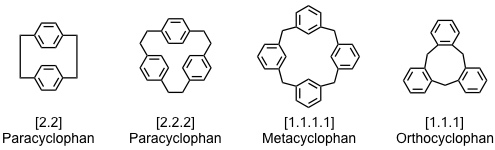
Die strukturelle Vielfalt von Rezeptoren auf Basis von Cyclophanen ist daher außerordentlich groß. Strukturell klassifiziert man Cyclophane in Abhängigkeit der:
- Anzahl der aromatischen Untereinheiten,
- der Stellung der Substituenten an diesen Untereinheiten
- und der Länge der Brücken zwischen ihnen.
Die wichtigen Rezeptorklassen der Calixarene und Resorcinarene basieren strukturell auf [1.1.1.1] Metacyclophanen. Sie werden in einem eigenen Kapitel vorgestellt. Rezeptoren auf Basis von [1.1.1] Orthocyclophanen bilden die eigene Rezeptorklasse der Cyclotriveratrylene.
Bindungseigenschaften
Neutrale aromatische Gastmoleküle binden an Cyclophane über dispersive Wechselwirkungen und π-π Wechselwirkungen. Bei der Komplexierung organischer Kationen leisten darüber hinaus Kation-π Wechselwirkungen entscheidende Beiträge.
Eine große Bedeutung in der Wirt-Gast Chemie von Cyclophanen haben wasserlösliche Derivate. Hier spielen bei der Komplexbildung hydrophobe Wechselwirkungen eine wichtige Rolle, d.h. die Freisetzung von Gastmolekülen aus den Solvathüllen von Wirt und Gast.
Das folgende Cyclophan ist in saurer Lösung aufgrund der Protonierung der Aminogruppen löslich. Es enthält als Bauelement zwei Diphenylmethanuntereinheiten, welche durch ihre Starrheit und definierte Krümmung dem Hohlraum des Rings eine charakteristische Form verleihen.
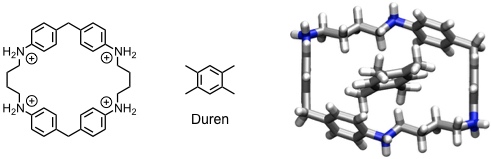
[Für eine interaktive Version der Kristallstruktur hier klicken]
Dieser Rezeptor bindet in wässriger Lösung einfache Aromaten. Die Kristallstruktur des Durenkomplexes zeigt, dass für die Substratbindung im Wesentlichen edge-to-face und face-to-face π-π Wechselwirkungen verantwortlich sind.
[Quelle: Odashima, K.; Itai A.; Iitake, Y.; Koga, K. J. Am. Chem. Soc. 1980, 102, 2504-2505: <doi>.]
Das Diphenylmethanbauelement findet man auch in einer Reihe weiterer Cyclophane, z. B. dem von F. Diederich entwickelten Derivat I. Diese Verbindung spielte eine wichtige Rolle bei der Charakterisierung von aromatischen Wechselwirkungen und der Untersuchung der thermodynamischen Beiträge von hydrophoben Wechselwirkungen in wässrigen Lösungsmitteln. Außerdem sind auf Basis dieses Cyclophans Enzymmodelle entwickelt worden.
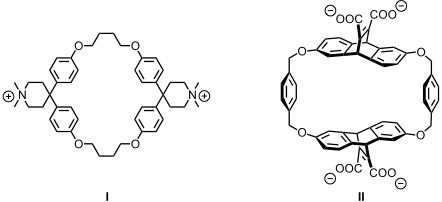
Mit dem in der Gruppe von D. A. Dougherty entwickelten Cyclophan II wurde die Bedeutung von Kation-π Wechselwirkungen bei Bindung kationischer Substrate an Rezeptoren mit aromatischen Untereinheiten untersucht.
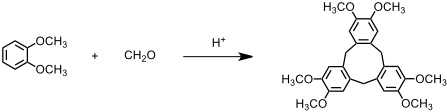
Cyclotriveratrylene
Derivate von [1.1.1] Orthocyclophanen, sogenannte Cyclotriveratrylene, entstehen bei der Umsetzung von Veratrol mit Formaldehyd im Sauren.
Die Eigenschaften dieser Verbindungen wurden vor allem in der Gruppe von A. Collet untersucht. Cyclotriveratrylen nimmt zwar bevorzugt eine schalenförmige Konformation an, die eine Einlagerung von Gastmolekülen erlaubt, die Bindungsaffinität ist aber wegen der Größe des Hohlraums und der Schwäche der Wechselwirkungen gering.
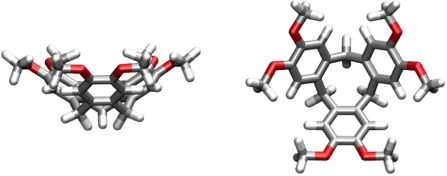
[Für eine interaktive Version der Kristallstruktur hier klicken]
Cryptophane
Zu interessanten Rezeptoren, den Cryptophanen, kommt man durch kovalente Verknüpfung zweier Cyclotriveratryleneinheiten. Dabei sind, je nachdem ob zwei enantiomere oder zwei identische Einheiten verknüpft werden, zwei Isomere möglich.
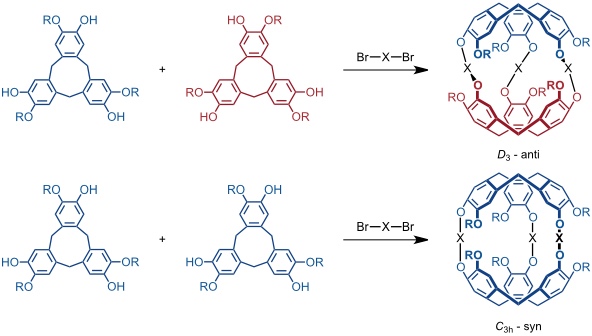
Während die direkte Verknüpfung zweier Cyclotriveratrylene meist nur in geringen Ausbeuten zum Erfolg führt, gelingt die Synthese meist besser, wenn man Cyclotriveratrylen zunächst mit geeigneten Substituenten versieht, welche in einem Folgeschritt cyclisiert werden können (Templatsynthese).
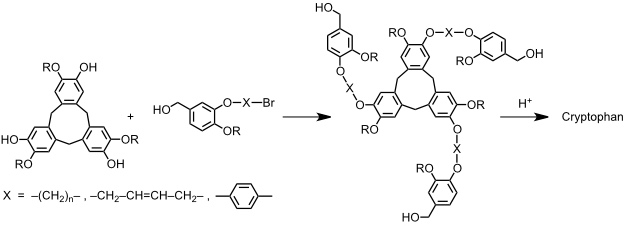
Die Bindungseigenschaften der Cryptophane können durch strukturell Variation der Linker zwischen den beiden Rezeptorhälften variiert werden. Cryptophane komplexieren Methan und halogenierte Kohlenwasserstoffen.
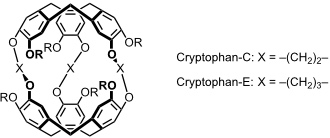
Dabei werden Gäste, die die Cryptophankavität optimal ausfüllen, besonders gut gebunden. Bei kleineren Gästen ist die Wechselwirkung schwächer und größere passen nicht in den Hohlraum. Cryptophan-C bindet dementsprechend CH2Cl2 am besten, während Cryptophan-E die höchste Bindungsaffinität für CHCl3 besitzt.
Dass die Komplementarität zwischen Größe des Gastes und Größe des zur Verfügung stehenden Hohlraums aber eine zu vereinfachte Erklärung für die beobachteten Selektivitäten ist, zeigt ein Vergleich der enthalpischen und entropischen Beiträge zur Stabilität der Komplexe zwischen Cryptophan-C bzw. Cryptophan-E mit CH2Cl2 und CHCl3.
| Cryptophan | Gast | ΔG0 / kJ mol-1 | ΔH0 / kJ mol-1 | ΔS0 / J K-1 mol-1 |
| C | CH2Cl2 | -15,1 | -16,3 | -4 |
| C | CHCl3 | -6,7 | -26,8 | -67 |
| E | CH2Cl2 | -11,7 | +4,2 | +25 |
| E | CHCl3 | -15,5 | -25,1 | -29 |
Die größere Affinität von Cryptophan-C für CH2Cl2 kann nach diesen Ergebnissen nicht auf einen besseren Fit des kleineren Gastes im Rezeptorhohlraum zurückzuführen sein, denn der enthalpische Beitrag zur Bindung von CHCl3 ist deutlich größer als der zur Bindung von CH2Cl2. Die Stabilität des Chloroformkomplex wird jedoch durch einen sehr ungünstigen Entropiebeitrag beeinträchtigt. Scheinbar füllt CHCl3 den Hohlraum des Cryptophans so gut aus, dass die Komplexbildung mit einem erheblichen Verlust von Konformationsfreiheitsgraden verbunden ist.
In dem größeren Cryptophan haben beide Gäste mehr Platz und in diesem Fall führt der günstigere Enthalpiebeitrag der Bindung von CHCl3 zu dem stabileren Komplex.
Die Einlagerung von Chloroform in das Innere von Cryptophan-E wurde mit Hilfe einer Kristallstruktur des Komplexes bestätigt.
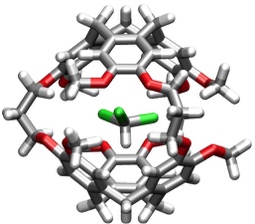
[Für eine interaktive Version der Kristallstruktur hier klicken]
[Quelle: Canceill, J.; Cesario, M.; Collet, A.; Guilhem, J.; Lacombe, L.; Lozach, B.; Pascard, C. Angew. Chem. 1989, 101, 1249-1251: <doi>.]
Neben neutralen Gastmolekülen werden auch organische Kationen in die Kavität von Cryptophanen eingelagert. So bindet das Tetramethylammoniumkation (NMe4+) an Cryptophan-E mit einer Assoziationskonstante von 2,5 · 105 M-1 (in CH2Cl2). Mit zunehmender Länge der Alkylgruppen an dem Ammoniumion sinkt die Komplexstabilität. Neben einer guten Größenkomplementarität ist in wichtiger Grund für die hohe Stabilität der Cryptophankomplexe organischer Kationen eine effiziente Beteiligung von Kation-π Wechselwirkungen.
Bücher
- Diederich, F. "Cyclophanes", RSC: Cambridge, 1991.
Literatur
- Collet, A. "Cyclotriveratrylenes and Cryptophanes" Tetrahedron 1987, 43, 5725-5759: <doi>.
- Collet, A.; Dutasta, J.-P.; Lozach, B.; Canceill, J. "Cyclotriveratrylenes and Cryptophanes: Their Applications to Host-Guest Chemistry and to the Design of New Materials" Top. Curr. Chem. 1993, 165, 103-130.
Calixarene
Struktur
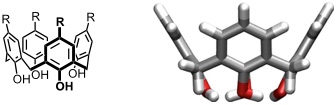
Calixarene sind [1n]Metacyclophane mit 4, 5, 6 oder mehr aromatische Untereinheiten.
Der Name Calixaren wurde von C. D. Gutsche eingeführt und beruht auf der Ähnlichkeit der (bei Calix[4]arenen thermodynamisch günstigsten) cone-Konformation mit einer griechischen Vase (griech. calix = vase).
Synthese
Calixarene werden durch Kondensation von Formaldehyd mit p-Alkylphenolen, zumeist 4-tert-Butylphenol, unter basischen Bedingungen dargestellt. Als Hauptprodukte fallen Calix[4]aren, Calix[6]aren und Calix[8]aren, also Oligomere mit gerader Anzahl der Untereinheiten an. Jedoch sind auch Calixarene mit ungerader Anzahl, z.B. Calix[5]aren, herstellbar.
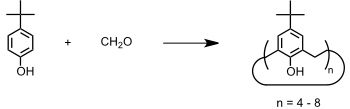
Als Basen werden NaOH oder KOH verwendet. Welches Calixaren jeweils bevorzugt gebildet wird, hängt von den Reaktionsbedingungen ab und es gibt optimierte Synthesebedingungen für jedes einzelne Calixaren. Z.T. werden Templateffekte für die bevorzugte Bildung eines Calixarens unter den jeweiligen Bedingungen verantwortlich gemacht.
Mechanismus
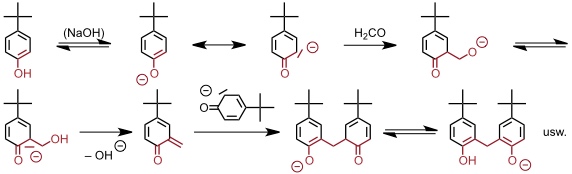
Derivatisierung
Calixarene sind vielseitig strukturell variierbar.
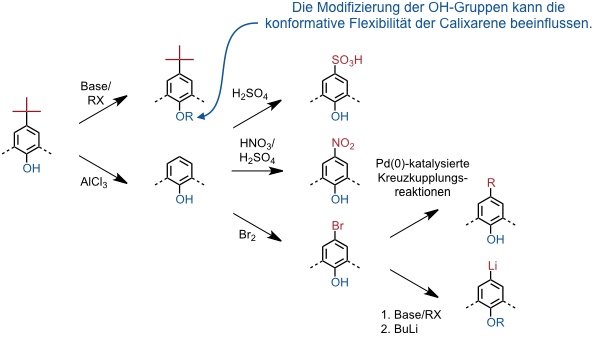
Es sind Synthesen entwickelt worden, um unsymmetrische Calixarene zu erhalten (eine oder zwei aromatische Untereinheiten haben andere Substituenten als die verbleibenden). Darüber hinaus gibt es auch eine Strategie zum sequentiellen Aufbau von Calixarenen, die die Darstellung von Verbindungen mit einer bestimmten Sequenz von Substituenten entlang des Rings erlaubt. Diese Synthesen sind aber komplex und zumeist wenig effizient.
Konformationen
Eine der wichtigsten Eigenschaften der Calixarene ist ihre konformative Beweglichkeit, die aus der mehr oder weniger freien Drehbarkeit um die σ-Bindung der Ar–CH2–Ar Gruppierung resultiert. Diese Flexibilität führt zu unterschiedlichen Konformationen, die man bei Calixarenen berücksichtigen muss. Bei Calix[4]aren existieren beispielsweise vier mögliche gegenseitige Anordnungen der Phenoluntereinheiten im Ring.
| cone | 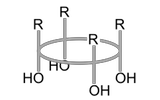 |  |
| partial cone |  |  |
| 1,2 alternate |  | 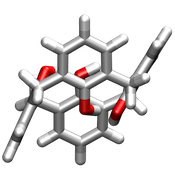 |
| 1,3 alternate |  | 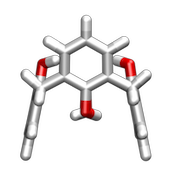 |
Bei größeren Calixarenen ist die Anzahl möglicher Konformere entsprechend größer. Das Calix[8]aren ist bereits so groß, dass man hierfür eine eingeschnürte Konformation diskutiert.
In den meisten Fällen ist die symmetrische cone-Konformation, die es bei allen Calixarenen gibt, für einen Einschluss von Gastmolekülen am besten geeignet.
Dies dürfte ein Grund sein, warum die cone-Konformation die thermodynamisch stabilste ist und in Lösung überwiegt. Bei Raumtemperatur findet jedoch eine schnelle Umwandlung der beiden entgegengesetzten cone-Konformationen statt. Dies kann man NMR spektroskopisch belegen, denn für die axialen und äquatorialen Protonen in den Methylenbrücken des Calix[4]arens wird im 1H-NMR Spektrum nur ein Singulett beobachtet. Aus der Temperaturabhängigkeit des Signals kann man eine freie Aktivierungsenthalpie für die Inversion von 68,6 kJ mol-1 (bei 25 °C in CDCl3) bestimmen. Bei Calix[8]aren ist der Wert vergleichbar (70,3 kJ mol-1). In polaren Lösungsmitteln sinkt die freie Aktivierungsenthalpie.
Auch methylierte Calixarenderivate besitzen geringere Aktivierungsenthalpien für die konformative Umwandlung.
Bei sukzessiver Alkylierung der OH-Gruppen steigt zunächst die Flexibilität der Calixarene, da auch andere Konformationen als die cone-Konformation populiert werden. Bei kleinen Alkylgruppen ist ein Durchschwingen der entsprechenden OR-Gruppen durch das Innere des Rings möglich, so dass letztlich flexible Makrocyclen erhalten werden. Ab einer Kettenlänge von drei C-Atomen (Propyl) werden die OR-Gruppen aber zu groß, um durch das Innere eines Calix[4]arenrings schwingen zu können. Bei Alkylierung der OH-Gruppen eines Calix[4]arens mit größeren Alkylgruppen werden daher meistens verschiedene Konformationen stabilisiert, welche voneinander getrennt werden können. Eine strukturelle Charakterisierung gelingt mit der 1H NMR Spektroskopie.
Bindungseigenschaften
Calixarene sind sehr vielseitige Wirtmoleküle. So wurden auf Basis von Calixarenen Wirte für kationische, anionische und neutrale Gastmoleküle entwickelt.

Je nach Art des Kations beruht die Bindung an Calixarene auf verschiedenen Wechselwirkungsarten. Darüber hinaus können bei einer Komplexbildung mit flexiblen Calixarenen unterschiedliche Konformationen stabilisiert werden.
 |  |  | |
| Kationen | Li+, Na+ | Ammoniumionen | Ag+ |
| Calixarenkonformation | cone | cone | partial cone |
| Wechselwirkungsart | Ion-Dipol | Kation-π | Ion-Dipol und Kation-π |
Die Einlagerung von Ammoniumionen in den Hohlraum von Calixarenen äußert sich in einem merklichen Hochfeldshift der Gastsignale im 1H NMR Spektrum. Anhand der Größenordnung der Shifts verschiedener Gastsignale kann man auf die bevorzugte Orientierung des Gastes innerhalb des Calixarenhohlraums schließen.
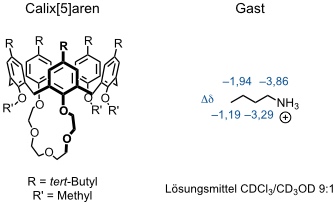
[Quelle: Pappalardo, S.; Parisi, M. F. J. Org. Chem. 1996, 61, 8724-8725: <doi>.]
Beispiele für Wirte für Metallkationen auf Basis von Calixarenen.
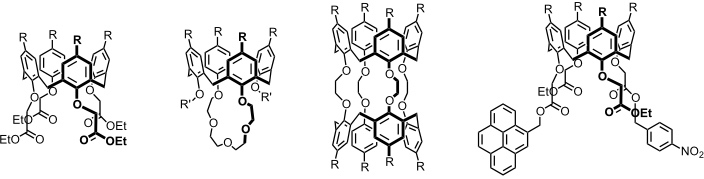
Bei dem letzten Derivat wird die Kationenbindung durch eine Veränderung des Fluoreszenzspektrums des Wirts signalisiert.
Wasserlösliche Calixarene mit Sulfonsäuregruppen komplexieren Ammoniumionen auch in Wasser, wofür im Wesentlichen elektrostatische Wechselwirkungen verantwortlich sind.
Für die Komplexbildung von Anionen oder neutralen Gastmolekülen mit Calixarenen existieren weniger Beispiele. Eine Anionenaffinität kann man z.B. durch Einführung von kationischen Gruppen oder Harnstoffsubstituenten in der Peripherie des Calixarenhohlraums induzieren.
Bücher
- Gutsche, C. D. "Calixarenes" RSC: Cambridge, 1989.
- Gutsche, C. D. "Calixarenes 2" RSC: Cambridge, 1997.
Literatur
Resorcinarene
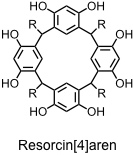
Struktur
Unter Resorcin[4]arenen versteht man die cyclischen Tetramere, die sich bei Reaktion von Resorcin (oder Pyrogallol) mit Aldehyden ergeben. Auch Resorcin[4]arene haben einen schalenförmigen Hohlraum. Dieser ist allerdings flacher mit einem größeren Durchmesser als der von Calixarenen.

Synthese
Resorcin[4]arene entstehen bei Kondensation von Aldehyden mit Recorcin. Im Gegensatz zur Synthese von Calixarenen wird die Reaktion aber unter sauren Bedingungen durchgeführt (HCl). Darüber hinaus werden mit Formaldehyd nur Polymere und keine cyclischen Produkte gebildet. Mit vielen anderen Aldehyden verläuft die Resorcin[4]arensynthese aber mit guten Ausbeuten.
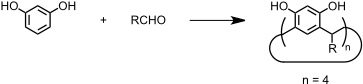
Die Methingruppen im Produkt, die die ursprünglichen Substituenten des verwendeten Aldehyds tragen, sind stereogene Zentren. Aus diesem Grund existieren für ein Resorcin[4]aren mit vier identischen Resten R insgesamt vier verschiedene Diastereomere. Für den Einsatz als Rezeptoren ist das C4-symmetrische rccc-Stereoisomer am besten geeignet und man hat Bedingungen gefunden, unter denen dieses Isomer bevorzugt gebildet wird.
Synthetisch leicht zugänglich ist nur das cyclische Tetramer, Resorcin[4]aren. Der Grund für die bevorzugte Bildung dieser Verbindung dürfte dessen Stabilisierung durch intramolekulare Wasserstoffbrücken zwischen den OH-Gruppen benachbarter aromatischer Untereinheiten sein.
Konformationen
Resorcin[4]aren ist konformativ ähnlich flexibel wie Calix[4]aren. Für die supramolekulare Chemie ist aber nur die C4v-symmetrische cone-Konformation von Bedeutung.
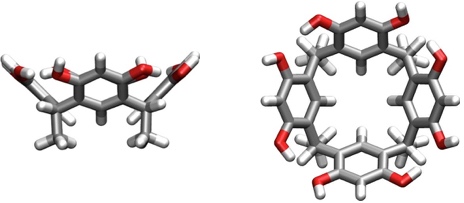
[Für eine interaktive Version der Kristallstruktur hier klicken]
Bindungseigenschaften
Resorcinarene binden in unpolaren, aprotischen Lösungsmitteln Diole, Dicarbonsäuren und Kohlenhydrate. Die wesentlichen Wechselwirkungen erfolgen über Wasserstoffbrücken, an denen jeweils zwei benachbarte OH-Gruppen des Wirtes und eine OH-Gruppe des Gastes beteiligt sind. Zudem sind C–H···π und Kation···π Wechselwirkungen möglich.
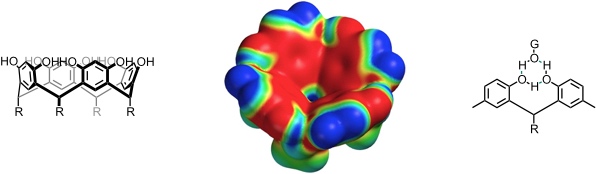
Durch ein Resorcin[4]aren lassen sich Monosaccharide, beispielsweise Methyl-β-D-glucopyranosid, in CCl4 solubilisieren, wobei man eine gewisse Selektivität in Abhängigkeit der Monosaccharidstruktur beobachtet.
Ursprünglich nahm man an, dass die Wechselwirkung zwischen Resorcin[4]aren und Methyl-β-D-glucopyranosid auf der Bildung eines 2:1 (Wirt:Gast) Komplexes beruht, in dem das Zuckermolekül zwischen zwei Resorcin[4]areneinheiten sandwichartig eingelagert ist.
Erst viel später stellte sich heraus, dass tatsächlich hochmolekulare Komplexe aus 6 Resorcin[4]aren und 3 Zuckermolekülen gebildet werden, die eine ähnliche Struktur besitzen, wie die im weiteren Verlauf des Kapitels vorgestellten Hexamere aus Resorcin[4]arenmolekülen. Für Glutarsäure wurde ein Aggregat aus 6 Resorcin[4]aren und 6 Glutarsäuremolekülen gefunden.
[Quellen: Tanaka, Y.; Ubukata, Y.; Aoyama, Y. Chem. Lett. 1989, 1905-1908: <doi>; Evan-Salem, T.; Baruch, I.; Avram, L.; Cohen, Y.; Palmer, L. C.; Rebek Jr., J. Proc. Natl. Acad. Sci. USA 2006, 103, 12296-12300: <doi>.]
Selbstaggregation
Resorcinarene kristallisieren aus geeigneten Lösungsmitteln und in Gegenwart geeigneter Gastmoleküle in Form von dimeren oder hexameren Aggregaten. In der unten abgebildeten Struktur werden die beiden Resorcinareneinheiten des Dimers durch acht cyclisch angeordnete Isopropanolmoleküle zusammengehalten. Der Hohlraum enthält fehlgeordnete Lösungsmittelmoleküle.
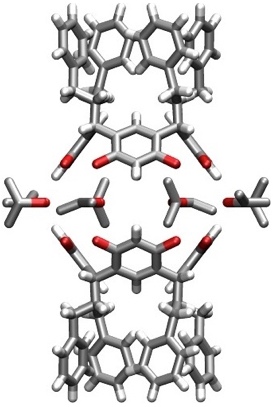
[Für eine interaktive Version der Kristallstruktur hier klicken]
[Quelle: Rose, K. N.; Barbour, L. J.; Orr, G. W.; Atwood, J. L. Chem. Commun. 1998, 407-408: <doi>.]
Dimere Aggregate, in denen der Hohlraum gefüllt ist, wurden ebenfalls gefunden. Ein Beispiel ist ein Dimer, in dem sich ein Tetramethylammoniumkation im Hohlraum befindet. Die beiden Resorcinareneinheiten werden durch Methanol, Wasser und Bromidionen zusammengehalten.
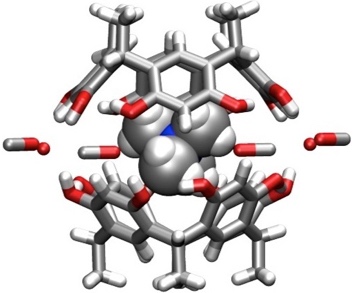
[Für eine interaktive Version der Kristallstruktur hier klicken]
[Quelle: Mansikkamäki, H.; Nissinen, M.; Schalley, C. A.; Rissanen, K. New. J. Chem. 2003, 27, 88-97: <doi>.]
Unter geeigneten Bedingungen kristallisieren Resorcinarene auch als Octahydrat eines Hexamers, in welchem die sechs Resorcin[4]areneinheiten die Flächen eines Würfels besetzen. Durch die schalenförmige Struktur des Resorcinarens führt dies insgesamt zu einer kugelförmigen Anordnung. Das Aggregat wird über 36 intermolekulare Wasserstoffbrücken zusammengehalten.
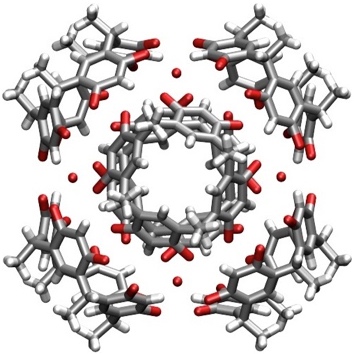
[Für eine interaktive Version der Kristallstruktur hier klicken]
[Quelle: MacGillivray, L. R.; Atwood, J. L. Nature 1997, 389, 469-472: <doi>.]
Analoge Strukturen besitzen die von Aoyama beschriebenen Kohlenhydratkomplexe von Resorcin[4]arenen, in denen OH Gruppen der Zuckerreste vermutlich Positionen der Wassermoleküle in dem hexameren Aggregat übernehmen.
Ein von Pyrogallol abgeleitetes Resorcin[4]aren (R = CH2CH(CH3)2) kristallisiert ebenfalls als Hexamer. In diesem Fall ist kein Wasser zur Überbrückung der einzelnen Untereinheiten nötig. Insgesamt stabilisieren 74 Wasserstoffbrücken dieses Aggregat. Der Hohlraum besitzt eine Größe von 1510 Å3.
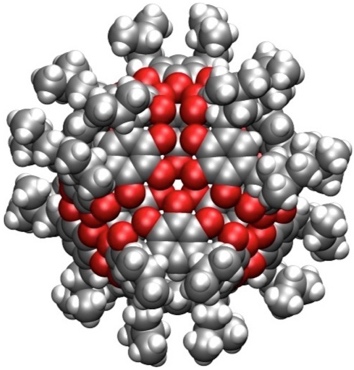
[Für eine interaktive Version der Kristallstruktur hier klicken]
[Quelle: Atwood, J. L.; Barbour, L. J.; Jerga, A. Chem. Commun., 2001, 2376-2377: <doi>.]
Literatur
- MacGillivray, L. R.; Atwood, J. L. "Strukturelle Klassifizierung von sphärischen molekularen Wirten und allgemeine Prinzipien für ihren Entwurf" Angew. Chem. 1999, 111, 1080-1096: <doi>.
- Rebek Jr., J. "Simultane Verkapselung: Moleküle unter sich" Angew. Chem. 2005, 117, 2104-2115: <doi>.
Vertiefte Cavitanden
Durch kovalente Verbrückung der OH-Gruppen zweier benachbarter aromatischer Untereinheiten kann der Hohlraum eines Resorcinarens vertieft werden. Solche Verbindungen wurden von D. J. Cram erstmals hergestellt und wegen ihres rigiden, strukturell definierten Hohlraums als Cavitanden bezeichnet.
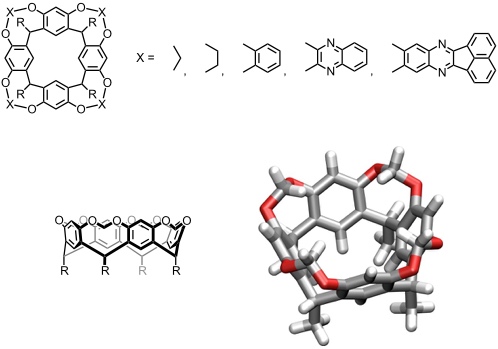
Der Begriff Cavitand ist aber nicht auf diese Verbindungsklasse beschränkt und wird im Allgemeinen für alle makrocyclischen Rezeptoren mit einer Kavität verwendet, in die ein Substrat eingelagert wird.
Sogenannte vertiefte Cavitanden (deep cavitands) enthalten aromatische Untereinheiten in der Peripherie. Solche Verbindungen sind flexibel und fluktuieren zwischen einer C4v und einer C2v symmetrischen Konformation.
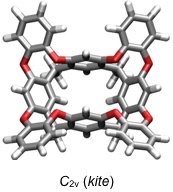
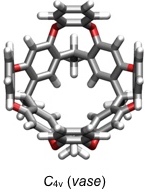
[Für interaktive Versionen der berechneten Strukturen auf die jeweilige Abbildung klicken]
Die C4v Konformation ist mit der cone Konformation von Calixarenen vergleichbar. Sie ist für die Substratbindung besser geeignet als die C2v Konformation.
Die C4v Konformation ist allerdings nur bei Temperaturen oberhalb von Raumtemperatur die stabilere, was entropische Gründe hat, denn sie wird wegen ihrer kleineren Oberfläche im Vergleich zur C2v Konformation von weniger Lösungsmittelmolekülen solvatisiert Die Anwesenheit geeigneter Gastmoleküle kann aber das Konformerengleichgewicht bei Raumtemperatur durch Komplexbildung auf die Seite der C4v symmetrischen Konformation verschieben (induced-fit).
Aufgrund der konformativen Flexibilität solcher Cavitanden ist das Gleichgewicht der Komplexbildung schnell auf der NMR Zeitskala.
Die C4v symmetrische Konformation eines Cavitanden kann durch geeignete Substituenten stabilisiert werden, welche die Seitenwände durch Ausbildung intramolekularer Wasserstoffbrücken zusammenhalten.
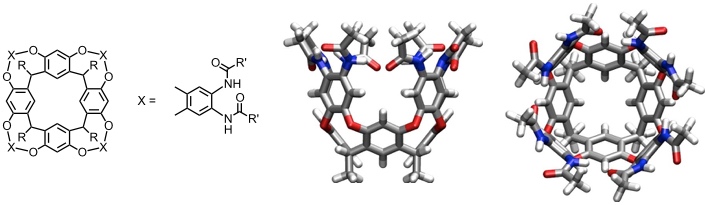
Solche Wirte bilden stabile Komplexe z.B. mit Adamantanderivaten. Interessanterweise ist der Gastaustausch in diesem Fall langsam auf der NMR Zeitskala. Man erklärt dies dadurch, dass beim Gastaustausch die Wasserstoffbrücken in der Peripherie des Cavitanden aufgebrochen werden müssen, was vor allem in unpolaren Lösungsmitteln ein energetisch ungünstiger Prozess ist. Ein gleichzeitiger Austausch des eingelagerten Gastmoleküls gegen Lösungsmittelmoleküle funktioniert aus Platzgründen nicht.
Diese self-folding Cavitands wurden in der Rebek Gruppe zur Entwicklung von "supramolekularen Reaktionsgefäßen" eingesetzt. So vermag ein Cavitand mit einer nach innen gerichteten Methylestergruppe Chinuclidin innerhalb von < 3 min bei Raumtemperatur zu methylieren.
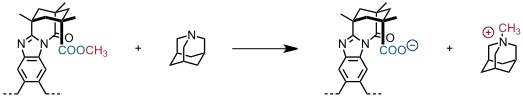
[Quelle: Purse, B. W.; Ballester, P.; Rebek Jr., J. J. Am. Chem. Soc. 2003, 125, 14682-14683: <doi>.]
Ein Cavitand, der am Rand der Öffnung einen Pyridonsubstituenten trägt, beschleunigt die Amidierung aktivierter Cholinester.
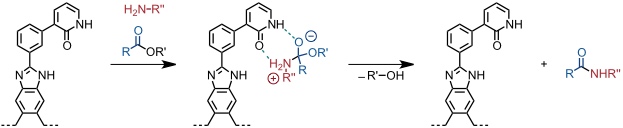
[Quelle: Gissot, A.; Rebek Jr., J. J. Am. Chem. Soc. 2004, 126, 7424-7425: <doi>.]
Literatur
- Rudkevich, D. M.; Rebek Jr., J. "Deepening Cavitands" Eur. J. Org. Chem. 1999, 1991-2005: <doi>.
Carceranden und Hemicarceranden
Der Begriff Carcerand wurde von D. J. Cram für sphärische Moleküle mit einem Hohlraum geprägt, in dem Gastmoleküle irreversibel komplexiert werden können. Die ersten Carceranden bestanden aus zwei verbrückten Resorcin[4]arenmolekülen mit Pyrogalloluntereinheiten.
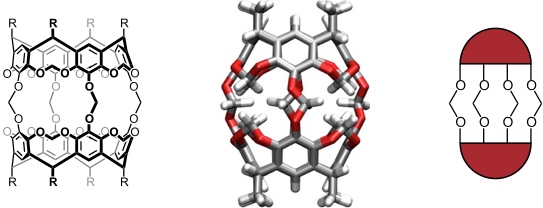
Sie wurden durch Reaktion der beiden Resorcin[4]arenuntereinheiten mit Bromchlormethan in DMF in Gegenwart einer Base (Kaliumcarbonat) synthetisiert. Berücksichtigt man, dass bei der Reaktion acht Bindungen auf die richtige Weise gebildet werden müssen, ist diese Synthese mit typischerweise ca. 60% Ausbeute überraschend effizient. Man vermutet, dass Templateffekte von Lösungsmittelmolekülen eine Rolle spielen, welche in dem Produkt permanent gebunden sind.
Schematische Darstellung des Templateffekts bei der Synthese von Carceranden
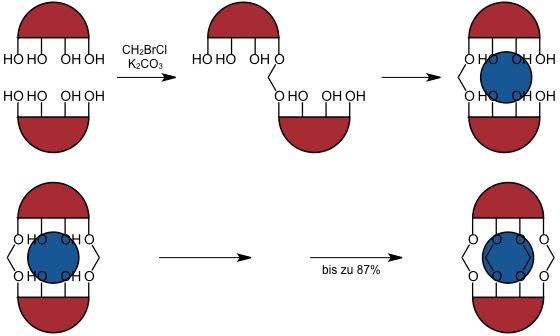
Als Template fungieren (bei einem Carcerand mit vier Methylenbrücken als Linker) typischerweise Lösungsmittelmoleküle wie Dimethylacetamid oder Dimethylformamid.
Unter Hemicarceranden versteht man Moleküle, bei denen zwischen den beiden Resorcinareneinheiten Lücken, existieren, die groß genug sind, damit ein Gastmolekül entweichen kann.
Hemicarceranden werden z.B. ausgehend von Resorcinarenen synthetisiert, die an einer aromatischen Untereinheit keine zusätzliche Hydroxygruppe tragen.
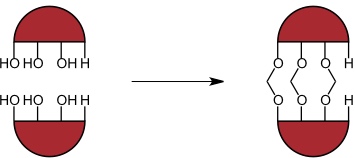
Eine Alternative zur Vergrößerung der Lücken zwischen den Resorcinarenuntereinheiten ist die Verwendung längerer Linker.
Hemicarceranden waren der Ausgangspunkt für die supramolekulare Stabilisierung reaktiver Intermediate, welche ansonsten in Lösung instabil sind und z.T. nur in der Argonmatrix beobachtet werden konnten.
So gelang es, durch Erhitzen einer Lösung in Chlorbenzol in Gegenwart eines Überschusses α-Pyron ein Molekül dieses Lacton in den Hohlraum eines Hemicarceranden mit drei Methylenbrücken einzuschließen. Bei Raumtemperatur ist dieser Komplex kinetisch inert, die Aktivierungsenergie für die Dekomplexierung ist also zu hoch. Durch Bestrahlen geht α-Pyron unter Abspaltung von CO2, das den Hemicarcerand verlassen kann, in Cyclobutadien über, welches bei Raumtemperatur NMR spektroskopisch untersucht werden konnte.
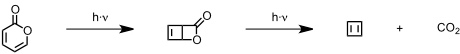
[Quelle: Cram, D. J.; Tanner, M. E.; Thomas, R. Angew. Chem. 1991, 103, 1048-1051: <doi>.]
Da Hemicarceranden nicht zulassen, dass ein eingeschlossenes Gastmolekül mit einem Reaktanden in Lösung reagiert, sind solche Untersuchungen zumeist auf intramolekulare photochemische Reaktionen beschränkt. Nur kleine Spezies, wie Protonen, Elektronen oder Wasser, können die Öffnungen von Hemicarceranden leicht passieren.
In Folgestudien konnten eine Reihe weiterer reaktiver Intermediate in solchen Hemicarceranden stabilisiert werden.
Arine
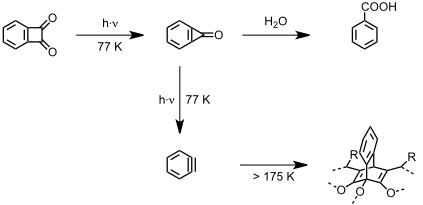
[Quelle: Warmuth, R. Angew. Chem. 1997, 109, 1406-1409: <doi>. Warmuth, R. Chem. Commun. 1998, 59-60: <doi>.]
1,2,4,6-Cycloheptatetraen, ein cyclisches Allen
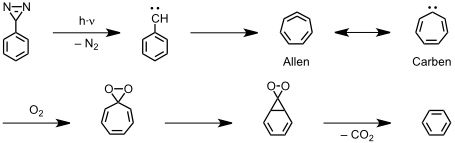
[Quelle: Warmuth, R.; Marvel, M. A. Angew. Chem. 2000, 112, 1168-1171: <doi>.]
Literatur
- Jasat, A.; Sherman, J. C. "Carceplexes and Hemicarceplexes" Chem. Rev. 1999, 99, 931-967: <doi>.
- Warmuth, R. "Inner-Phase Stabilization of Reactive Intermediates" Eur. J. Org. Chem. 2001, 423-437: <doi>.
- Warmuth, R.; Yoon, Y. "Recent Highlights in Hemicarcerand Chemistry" Acc. Chem. Res. 2001, 34, 95-105: <doi>.
- Sherman, J. "Molecules That Can't Resist Templation" Chem. Commun. 2003, 1617-1623: <doi>.
Pillararene
Struktur
Bei der erst 2008 von T. Ogoshi beschriebenen Rezeptorklasse der Pillararene handelt es sich um Makrocyclen mit 1,4- Hydrochinoneinheiten, die in 2,5-Position über Methylenbrücken verknüpft sind. Pillararene sind also eng verwandt mit Calixarenen oder Resorcinarenen, aufgrund der Verknüpfung der Untereinheiten in para- anstelle in meta-Position unterscheiden sich die Konformationen von Pillararenen signifikant von denen der anderen beiden Cyclophantypen.
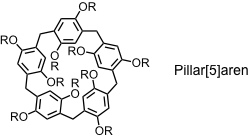
Das am leichtesten zugängliche Pillararen enthält 5 Hydrochinonuntereinheiten, aber auch größere Pillar[6]arene sowie Pillar[7]arene sind bekannt.
Im Gegensatz zu Calixarenen oder Resorcinarenen haben Pillararene keine schalenförmige, sondern eine zylindrische Struktur.
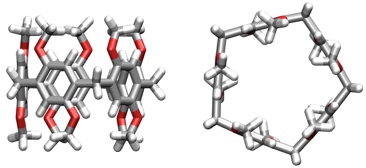
[Für eine interaktive Version der Kristallstruktur hier klicken]
Synthese
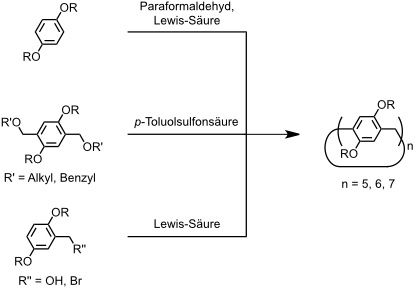
Zur Synthese von Pillararenen wurden bisher drei Strategien entwickelt:
- die Lewis-Säure (z.B. AlBr3) katalysierte Kondensation von 1,4-Dialkoxybenzolderivaten,
- die Kondensation von 1,4-Dialkyoxy-2,5-bis(alkoxymethyl)benzolderivaten in Gegenwart von p-Toluolsulfonsäure.
- die von Lewis-Säuren vermittelte Cyclooligomerisierung von 2,5-Dialkoxybenzylakoholen oder -bromiden.
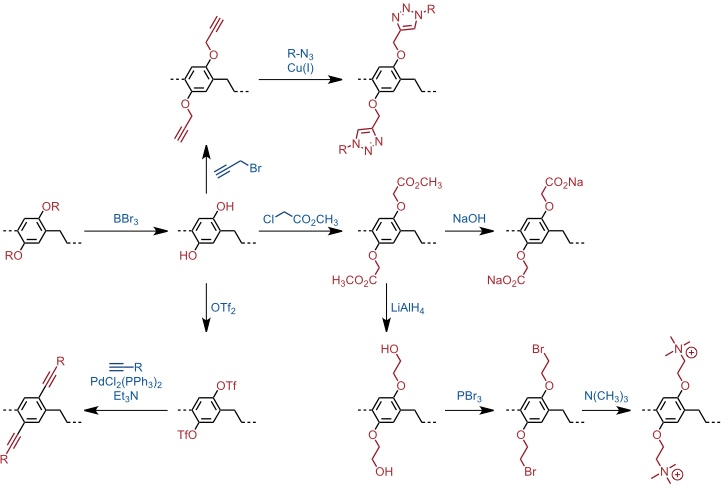
Derivatisierung
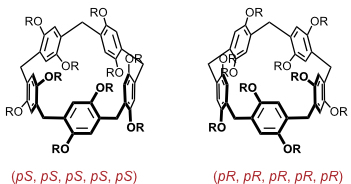
Konformationen
Pillararene sind konformativ flexibel. Die wichtigste konformative Flexibilität beruht auf der Rotation der aromatischen Untereinheiten, durch die sich die Orientierungen der OR-Gruppen entlang des Rings verändern. Insgesamt existieren aus diesem Grunde für Pillar[5]aren acht verschiedene stereoisomere Konformationen. Aufgrund der planaren Chiralität von Pillararenen verhalten sich diese Stereoisomere paarweise enantiomer zueinander. Dies ist am Beispiel der beiden C5-symmetrischen Konformationen in der Abbildung gezeigt.
Eine Rotation der aromatischen Untereinheiten kann durch Einführung sterisch anspruchsvoller Substituenten OR- Gruppen verhindert werden. Sogar mit R = Dodecyl wird aber noch eine Rotation beobachtet. Erst mit R = Cyclohexylmethyl werden die Konformationen der Pillararene stabilisiert.
Bindungseigenschaften
Der Hohlraum von Pillar[5]aren hat einen Durchmesser von ca. 5 Å, seine Dimensionen ähneln daher den Hohlräumen von α-Cyclodextrin oder Cucurbit[6]uril. Damit ist Pillar[5]aren zur Bindung linearer Alkane oder von Aromaten gut geeignet.
Das elektrostatische Potential der elektronenreichen aromatischen Untereinheiten ist stark negativ. Dementsprechend ist eine Gastbindung über Kation-π oder CH-π Wechselwirkungen bevorzugt.
Bindungseigenschaften in organischen Lösungsmitteln
In organischen Lösungsmitteln binden Pillararene halogenierte Alkane oder kationische Substrate, beispielsweise Ammoniumionen oder Pyridiniumsalze.
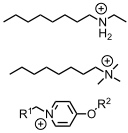
Bindungseigenschaften in Wasser
Anionische wasserlösliche Pillararene binden Kationen, z.B. Paraquatderivate, während kationische Pillararene organische Anionen komplexieren, z.B. Alkylsulfonate.
Es wurden auch verbrückte Bis(pillararene) dargestellt. Außerdem wurden Pillararene zum Aufbau von Rotaxanen oder Polyrotaxanen und für andere Anwendungen verwendet.
Literatur
- Cragg, P. J.; Sharma, K. "Pillar[5]arenes: fascinating cyclophanes with a bright future" Chem. Soc. Rev. 2012, 41, 597-607: <doi>.
- Xue, M.; Yang, Y.; Chi, X.; Zhang, Z.; Huang, F. "Pillararenes, a new class of macrocycles for supramolecular chemistry" Acc. Chem. Res. 2012, 45, 1294-1308: <doi>.
- Ogoshi, T.; Yamagishi, T. "New synthetic host pillararenes: their synthesis and application to supramolecular materials" Bull. Chem. Soc. Jpn. 2013, 86, 312-332: <doi>.
- Ogoshi, T.; Yamagishi, T. "Pillar[5]- and pillar[6]arene-based supramolecular assemblies built by using their cavity-size-dependent host-guest interactions" Chem. Commun. 2014, 50, 4776-4787: <doi>.
Cucurbiturile
Struktur
Bereits 1905 berichtete Behrend und Mitarbeiter von der Universität Hannover über die Produkte der Kondensation von Glycoluril und Formaldehyd (Behrends Polymer). Man erhielt nach Umkristallisation aus konzentrierter Salzsäure eine kristalline Substanz, deren Struktur erst 1981 von Mock und Mitarbeitern aufgeklärt wurde. Es handelt sich um einen Makrocyclus mit sechs verbrückten Glycolurileinheiten. Aufgrund der Ähnlichkeit der molekularen Struktur dieser Verbindung mit einem Kürbis (zur Cucurbitaceae-Familie gehörig), gab man dieser Verbindung den Namen Cucurbituril. Inzwischen sind Homologe bekannt. Zur Klassifizierung gibt man darum die Ringgröße von Cucurbiturilen ihm Namen, in Anlehnung an die bei Calixarenen gebräuchliche Nomenklatur, in eckigen Klammern an (Cucurbit[n]uril).
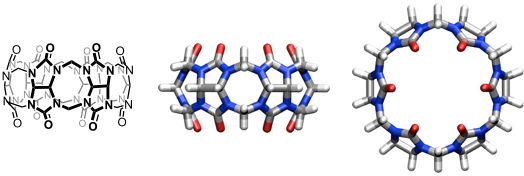
[Für eine interaktive Version der Kristallstruktur hier klicken]
[Quelle: Behrend, R.; Meyer, E.; Rusche, F. Liebigs Ann. Chem. 1905, 339, 1-37: <doi>.]
Die Familie der Cucurbiturile umfasst heutzutage die Ringgrößen zwischen 5 und 10, wobei sich die molekularen Dimensionen der einzelnen Vertreter charakteristisch unterscheiden.
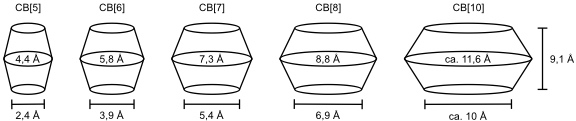
Synthese
Bei der von Behrend und Mock verwendeten Synthese wurde ausschließlich die Bildung von Cucurbit[6]uril beobachtet. Erst später wurden Reaktionsbedingungen entwickelt, die auch die Bildung von Cucurbiturilen mit anderen Ringgrößen erlauben. Alle Synthesemethoden basieren auf der Kondensation von Glycoluril mit Formaldehyd unter sauren Bedingungen.
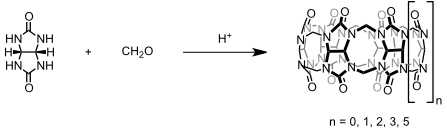
Durch Einsatz von substituierten Glycolurilderivaten können Cucurbiturile mit unterschiedlichen Substituenten in der Peripherie erhalten werden. Die Darstellung dieser Verbindungen erfordert oft eine sorgfältige Anpassung der Reaktionsbedingungen bzw. sogar andere synthetische Strategien.
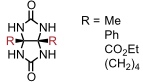
Cucurbiturile mit R = Me können oxidativ mit K2S2O8 (Kaliumperoxodisulfat) in die entsprechenden perhydroxylierten Verbindungen (R = OH) überführt wird, was einen Zugang zu weiteren Derivaten eröffnet.
Ein Nachteil von Cucurbiturilen mit R = H ist ihre relativ geringe Löslichkeit in Wasser oder organischen Lösungsmitteln: CB[6] und CB[8] sind praktisch unlöslich in Wasser, während CB[5] und CB[7] mäßig löslich sind. Cucurbiturile sind im Allgemeinen schlechter wasserlöslich als Cyclodextrine. In Salzlösungen steigt die Löslichkeit aufgrund der Bildung von Komplexen mit den Metallionen, welche an die C=O Gruppen der Cucurbiturile koordinieren, an.
Interessanterweise sind die Cyclohexylderivate (R = (CH2)4) von Cucurbit[6]uril und Cucurbit[5]uril wesentlich besser löslich in Wasser (200 mM) und organischen Lösungsmitteln (bis zu 20 mM in MeOH, DMSO, DMF oder CH3CN) als die entsprechenden Derivate mit R = H.
Konformationen
Die kleineren Cucurbiturile sind sehr starr und damit sehr gut für die Gastbindung vororganisiert. Dies erklärt z.T. die extrem hohen Stabilitätskonstanten, die für einige Cucurbiturilkomplexe beobachtet wurden. Bei größeren Cucurbiturilen kann der Ring leicht deformiert werden und beispielsweise eine eher elliptische Form annehmen.
Bindungseigenschaften
Das elektrostatische Potential von Cucurbiturilen ist an den Eingangsöffnungen und im Innenraum der Kavität wesentlich negativer als beispielsweise bei Cyclodextrinen. Dies erklärt, warum Cucurbiturile bevorzugt kationische Gäste binden, während Cyclodextrine bevorzugt mit neutralen oder anionischen Gästen wechselwirken.
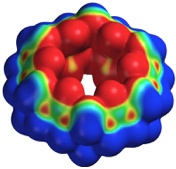
Cucurbiturile wechselwirken mit Metallkationen und insbesondere mit Kationen organischer Verbindungen, insbesondere Ammoniumionen. Dabei wird die Wechselwirkung mit Ammoniumionen auch zum Aufbau komplexerer Strukturen (Rotaxane, selbstassoziierte Systeme) ausgenutzt.
Bindung von Metallionen
log Ka Werte der Bindung von Metallionen an Cucurbit[6]uril in HCO2H/ H2O 1:1 und an 18-Krone-6 in Wasser bei 25 °C
| Li+ | Na+ | K+ | Rb+ | Ca2+ | Sr2+ | Ba2+ | |
| CB[6] | 2,38 | 3,23 | 2,79 | 2,68 | 2,80 | 3,18 | 2,83 |
| [18]Krone-6 | - | 0,80 | 2,03 | 1,56 | < 0,5 | 2,72 | 3,87 |
Die geringe Selektivität und das Fehlen eines eindeutigen Trends in den log Ka Werten wurde auf die mangelnde Kompatibilität der Kationenradien mit dem Innenradius des starren Cucurbit[6]uril Moleküls zurückgeführt.
In Kristallstrukuren wurde die Koordination von Metallionen (z.B. Na+) an die Carbonylgruppen der Cucurbiturile beobachtet. In hochkonzentrierten Salzlösungen nimmt man solche Komplexe auch in Lösung an. Sie erklären die verbesserte Löslichkeit von Cucurbiturilen unter diesen Bedingungen.
Bindung von Ammoniumionen
Bindung von Aminen und Diaminen an Cucurbit[6]uril in HCO2H/H2O 1:1
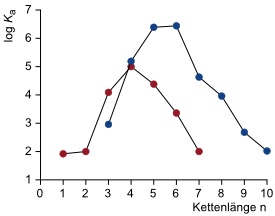
Man beobachtet eine klare Kettenlängenabhängigkeit der Komplexstabilität. Bei den Monoaminen (rote Punkte) sind jene Komplexe am stabilsten, bei denen das Gastmolekül vollständig in das Cucurbituril eingeschlossen ist. Bei den Diaminen (blaue Punkte) ist eine Kettenlänge am günstigsten, die es beiden Ammoniumgruppen erlaubt, mit den Carbonylgruppen an den Hohlraumöffnungen des Cucurbit[6]urils zu wechselwirken. Zu beachten sind außerdem die extrem hohen Stabilitäten mancher Diaminkomplexe, die log Ka Werte von fast 7 erreichen.
[Quelle: Mock, W. L.; Shih, N. Y. J. Org. Chem. 1986, 51, 4440-4446: <doi>.]
Ultrahohe Affinitäten
Extrem hohe Stabilitäten wurden für einige Komplexe des Cucurbit[7|urils mit kationischen Gästen beobachtet.
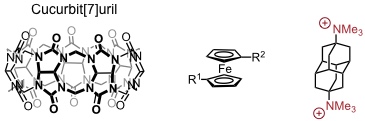
Bindung von Ferrocenderivaten an Cucurbit[7]uril in Wasser (Gegenionen: Iodid)
| R1 | R2 | Ka / M-1 | ΔH0 / kJ mol-1 | TΔS0 / kJ mol-1 |
| –CH2OH | –H | 3,2 × 109 | -90 | -36 |
| –CH2N+(CH3)3 | –H | 4,1 × 1012 | -89 | -17 |
| –CH2N+(CH3)3 | –CH2N+(CH3)3 | 3.0 × 1015 | -92 | -4 |
Die Stabilität dieser Komplexe erreicht oder überschreitet sogar diejenige des Biotin-Streptavidin Komplexes (1013,4 M-1). Für das oben dargestellte maßgeschneidertes Diammoniumion wurde sogar kürzlich eine Bindungskonstante Ka von 7,2 × 1017 M-1 in D2O bestimmt.
Als Ursache für die hohe Stabilität wird die Starrheit des Cucurbit[7]urilrings (und der Gäste) gesehen. Bei der Gastbindung werden wenige Freiheitsgrade eingeschränkt, sodass die Bildung des Komplexes entropisch nicht stark benachteiligt ist (und diesbezüglich von der Desolvatation des Wirtes und des Gastes profitiert). Die Komplexstabilität wird wesentlich von der sehr günstigen Komplexbindungsenthalpie geprägt, die interessanterweise von der Struktur der Gäste recht unabhängig ist.
[Quellen: Rekharsky, M. V.; Mori, T.; Yang, C.; Ko, Y. H.; Selvapalam, N.: Kim, H.; Sobransingh, D.; Kaifer, A. E.; Liu, S.; Isaacs, L.; Chen, W.; Moghaddam, S.; Gilson, M. K.; Kim, K.; Inoue, Y. Proc. Natl. Acad. Sci. USA 2007, 104, 20737-20742: <doi>. Moghaddam, S.; Inoue, Y.; Gilson, M. K. J. Am. Chem. Soc. 2009, 131, 4012-4021: <doi>. Cao, L.; Šekutor, M.; Zavalij, P. Y.; Mlinaric-Majerski, K.; Glaser, R.; Isaacs, L. Angew. Chem. 2014, 126, 1006-1011: <doi>.]
High-Energy Wasser
Während die günstige Komplexbildungsenthalipe von einigen Autoren auf die Stärke der Wechselwirkungen zwischen dem Cucurbituril und dem Gast zurückgeführt wird, zeigen aktuellere Untersuchungen, dass die Freisetzung energiereichen Wassers hierfür wahrscheinlich eher verantwortlich ist.
Moleküldynamiksimulationen zeigen, dass Wassercluster in einem hydrophoben Hohlraum nicht die optimale Anzahl intermolekularer Wasserstoffbrücken ausbilden können.
Simulation von Kavitätswasser in Cucurbiturilen
| System | N | m | Z |
| flüssiges Wasser | 3,62 | ||
| CB6 | 3,3 | 1,71 | 6,3 |
| CB7 | 7,9 | 2,52 | 8,7 |
| CB8 | 13,1 | 3,06 | 7,3 |
N = Durchschnittliche Zahl der Kavitätswassermoleküle
m = Durchschnittliche Zahl der Wasserstoffbrücken pro gebundenem Kavitätmolekül
Z = N (3,62 - m)
Damit ist Z ein relatives Maß für den enthalpischen Gewinn der Freisetzung von gebundenen Wassermoleküle. Er berücksichtigt die Anzahl der gebundenen Wassermoleküle und das Wasserstoffbrückendefizit jedes einzelnen Wassermoleküls bezogen auf die Situation in flüssigem Wasser (3,62 - m).
Die Betrachtung zeigt ein Maximum von Z für Cucurbit[7]uril und vergleichsweise große Werte für alle Cucurbiturile. Bei anderen Wirten liegt Z typischerweise zwischen 1,2 und 3,6 (z.B. Calix[4]aren = 1,4). Damit profitiert die Komplexbildung bei Cucurbituril in besonderem Maße von der Freisetzung des high-energy Wassers.
[Quelle: Biedermann, F.; Nau, W. M.; Schneider, H.-J. Angew. Chem. 2014, 126, 11338-11352: <doi>.]
Bambusurile
In einem Makrocyclus, der sechs Glycourileinheiten über jeweils nur eine Methylenbrücke verbrückt enthält, weisen die Methinprotonen der Glycolurileinheiten nach innen. Das elektrostatische Potential dieses Makrocyclus ist innen positiv, weswegen diese Verbindung Anionen bindet. Aufgrund ihrer molekularen Struktur wird sie als Bambusuril bezeichnet.
Aufgrund der Größe des Hohlraums bindet Bambusuril am stärksten an Iodidanionen. Die Stabilitätskonstante des Iodidkomplexes in D2O/CD3CN (1:1) beträgt 8.9 × 105 M-1.
[Für eine interaktive Version der Kristallstruktur hier klicken]
[Quelle: Svec, J.; Necas, M.; Sindelar, V. Angew. Chem. 2010, 122, 2428-2431: <doi>. Svec, J.; Dusek, M.; Fejfarova, K.; Stacko, P.; Klán, P.; Kaifer, A. E.; Li, W.; Hudeckova, E.; Sindelar, V. Chem. Eur. J. 2011, 17, 5605-5612: <doi>.]
Literatur
- Lagona, J.; Mukhopadhyay, P.; Chakrabarti, S.; lsaacs L. "Die Cucurbit[n]uril-Familie" Angew. Chem. 2005, 117, 4922-4949: <doi>.
- Kim, K.; Selvapalam, N.; Ko, Y. H.; Park, K. M.; Kim, D.; Kim, J. "Functionalized Cucurbiturils and Their Applications" Chem. Soc. Rev. 2007, 36, 267-279: <doi>.
- Masson, E.; Ling, X.; Joseph, R.; Kyeremeh-Mensah, L.; Lu, X. "Cucurbituril Chemistry: A Tale of Supramolecular Success" RSC Advances 2012, 2, 1213-1247: <doi>.
Clefts
Definition
Unter den Begriffen "Molecular Clefts" und "Pinzettenmolekülen" fasst man synthetische Rezeptoren zusammen, die nicht-cyclisch sind, aufgrund einer starren Struktur jedoch konvergierende funktionelle Gruppen besitzen, zwischen denen ein Gast eingelagert werden kann. Der Begriff "Molecular Cleft" wurde ursprünglich von J. Rebek Jr. geprägt. Man versteht darunter Verbindungen, bei denen die Bindung des Substrats in einer Spalte oder Vertiefung des Rezeptors erfolgt.
Während der Begriff "Molecular Cleft" heute selten für nicht-cyclische Rezeptoren verwendet wird, hat sich der Begriff Pinzettenmolekül ("Molecular Tweezer") durchgesetzt. Er wird aber weiter gefasst und man versteht darunter jede Art von nicht-cyclischen Rezeptoren mit konvergierenden funktionellen Gruppen.
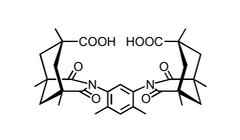
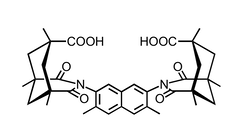
Die von Rebek entwickelten Clefts basieren auf der Kemp'sche Säure. Die Methylgruppen am Cyclohexanring nehmen in dieser Verbindung äquatoriale Stellungen ein, sodass die drei Carboxylgruppen konvergieren.
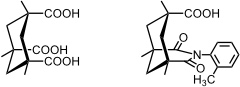
Durch Kondensation der Kemp'schen Säure mit einem aromatischen Amin erhält man ein Imid, in dem die verbleibende Carboxylgruppe parallel zum Substituenten des Imids angeordnet ist. Die Rotation um die C–N Bindung kann durch eine Methylgruppe in ortho-Position verhindert werden. Die Reaktion mit aromatischen Diaminen liefert entsprechend nicht-cyclische Rezeptoren, in denen die beiden Carboxylgruppen für die Substratbindung vororientiert sind.
 |  |
 |  |
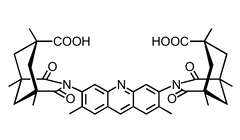 | 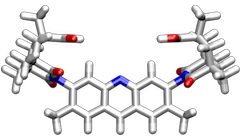 |
[Für interaktive Versionen der berechneten Strukturen auf die jeweilige Abbildung klicken]
Der Abstand der beiden Carboxylgruppen wird durch Variation der Struktur des Linkers kontrolliert.
Solche Clefts können mit verschiedenen Substraten wechselwirken:
- Der kleinste Rezeptor bindet in Form des Dicarboxylats zweiwertige Metallkationen wie Ca2+ oder Mg2+.
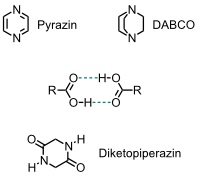
- Der größte Rezeptor bindet in Chloroform Diamine wie Pyrazin oder DABCO.
- Bei der Bindung von Aminosäuren bindet eine Carboxylgruppe des Rezeptors an die Carboxylgruppe des Substrats. Zusätzlich erfolgt Salzbildung zwischen der anderen Carboxylgruppe des Rezeptors und der Aminogruppe des Gastes.
- Dicarbonsäuren werden über Wasserstoffbrücken gebunden, die denen ähneln, welche man in Carbonsäuredimeren findet.
- Neutrale Substrate wie Diketopiperazine können ebenfalls über Wasserstoffbrücken mit dem größten Rezeptor wechselwirken.
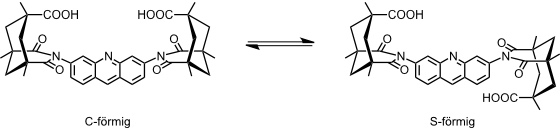
Derivate der Rezeptoren ohne die Methylgruppen an den aromatischen Linkern sind konformativ flexibel und können eine C-förmige und eine S-förmige Anordnung einnehmen. Bei Anwesenheit geeigneter Gastmoleküle wird der Rezeptor aber in einer C-förmigen Anordnung fixiert, da nur in dieser eine gleichzeitige Bindung beider Carboxylatgruppen an das eingelagerte Gastmolekül möglich ist. Dieser Mechanismus der Komplexbildung kann als "induced-fit" bezeichnet werden.
Die Komplexierung von Nucleobasen, z.B. Adenin, erlauben Derivate der Clefts, in denen zwei Carboxylgruppen der Kemp‘schen Säure in ein unsubstituiertes Imid überführt wurden. Die verbleibende Carboxylgruppe wurde mit aromatischen Aminen gekuppelt. Diese Verbindungen binden Thymin durch Watson-Crick-Basenpaarung mit dem Imid. Eine zusätzliche Stabilisierung des Komplexes erfolgt durch π-π-Wechselwirkungen zwischen dem Substrat und dem parallel angeordneten aromatischen Substituenten des Rezeptors.
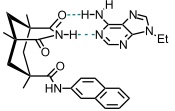
Die Wechselwirkung solcher Imide mit Amiden wurde in der licht-getriebenen enantioselektiven Organokatalyse für eine intramolekulare [2+2]-Cycloaddition ausgenutzt.
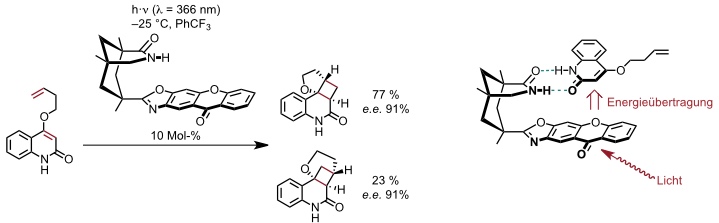
[Quelle: Müller, C.; Bauer, A.; Bach, T. Angew. Chem. 2009, 121, 6767-6769: <doi>.]
Eine Cleft zur Bindung von Ethyladenin stellt folgender Rezeptor dar. Er bindet den Gast mit einer Affinität von ca. 105 M-1 in Chloroform.
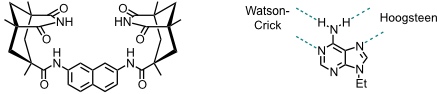
[Quelle: Benzing, T.; Tjivikua, T.; Wolfe, J.; Rebek Jr., J. Science 1988, 242, 266-268: <doi>.]
Literatur
Pinzettenmoleküle
Definition
Unter dem Begriff Pinzettenmolekül, der von H. W. Whitlock eingeführt wurde, versteht man Rezeptoren mit zwei parallel angeordneten aromatischen Untereinheiten, zwischen denen der Gast gebunden wird. Die von Zimmerman entwickelten Rezeptoren entsprechen strukturell dieser von H. W. Whitlock eingeführten Definition, d.h. sie enthalten zwei parallel angeordnete aromatische Untereinheiten.
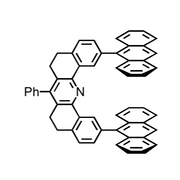 | 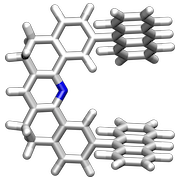 |
[Für interaktive Versionen der berechneten Strukturen auf die jeweilige Abbildung klicken]
Diese Rezeptoren sind für die Interkalation aromatischer Gäste zwischen den beiden Anthraceneinheiten vororganisiert. Allerdings ist der Abstand der Anthracenreste - wenn diese im 90° Winkel zum Acridin angeordnet sind - mit 7,2 Å für ein effizientes π-Stacking etwas zu groß. Durch eine Verringerung des Winkels auf 60° verkleinert sich ihr Abstand jedoch auf 6,3 Å, wodurch sich das System den Erfordernissen eines Komplexes mit einem Aromaten anpassen kann.
Für die beiden dargestellten Systeme wurde eine Wechselwirkung mit elektronenarmen Aromaten nachgewiesen.
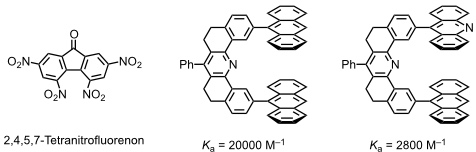
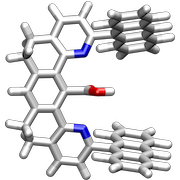
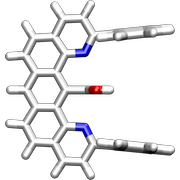
Die nächste Generation Pinzettenmoleküle enthielt zwischen den Anthracenresten eine Carboxylgruppe, die mit geeigneten Gastmolekülen über Wasserstoffbrücken wechselwirken kann.
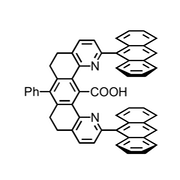 | 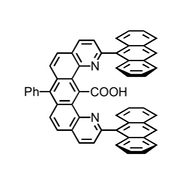 |
 |  |
[Für interaktive Versionen der berechneten Strukturen auf die jeweilige Abbildung klicken]
Diese Rezeptoren bilden mit 9-Propyladenin Komplexe in CDCl3.
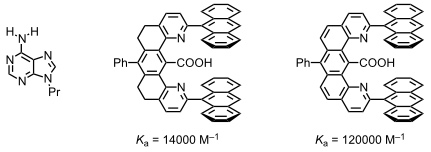
Die Wechselwirkung einer Carboxylgruppe mit 9-Propyladenin kann auf zweierlei Weise erfolgen.
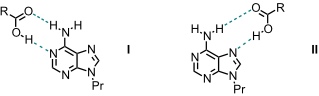
Geometrie I ähnelt einer Watson-Crick- und Geometrie II einer Hoogsteen-Basenpaarung. Untersuchungen mit Modellverbindungen haben ergeben, dass das Substrat bevorzugt mit Geometrie II gebunden wird. So wird ein monomethyliertes Adenin mit ähnlicher Stärke gebunden wie unsubstituiertes Adenin. Da die Methylgruppe in dem monomethylierten Gast bevorzugt eine Orientierung annimmt, die Geometrie I verhindert, ist dies ein starker Hinweis darauf, dass Geometrie II überwiegt.

Weitere Untersuchungen bestätigten dieses Ergebnis. Als Ursache für die Bildung des Komplexes II wird angenommen, dass bei dieser Geometrie die Oberflächen der in Kontakt stehenden aromatischen Systeme größer sind.
Literatur
- Zimmerman, S. C. "Rigid Molecular Tweezers as Hosts for the Complexation of Neutral Guests" Top. Curr. Chem. 1993, 165, 71-102.
Letzte Änderung: 23-03-30. Email