Lehre - Supramolekulare Chemie - Intermolekulare Wechselwirkungen
Thermodynamik
Thermodynamische Aspekte einer Komplexbildung
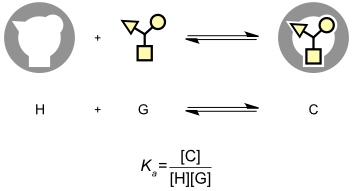
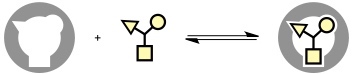
Die Bildung eines Komplexes aus einem synthetischen Rezeptor und seinem Substrat kann man als Gleichgewichtsreaktion beschreiben. Damit kann diese Reaktion durch das Massenwirkungsgesetz beschrieben werden. Die Lage des Gleichgewichts ergibt sich aus der Gleichgewichtskonstante oder Stabilitätskonstante Ka, die ein Maß für die Stabilität des Komplexes darstellt. Je größer Ka desto stabiler ist der Komplex. Ka hat bei 1:1 Komplexen die Dimension einer inversen Konzentration.
Manchmal wird anstelle von Ka die Dissoziationskonstante Kd angegeben, die den Kehrwert von Ka darstellt. Kd hat die Konzentration einer Dimension und wird um so kleiner, je höher die Komplexstabilität ist.
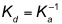
Ka steht gemäß folgender Beziehungen mit den thermodynamischen Größen der Komplexbildung in Zusammenhang.

Die Stabilität eines supramolekularen Komplexes ist im Wesentlichen von der Art der zugrunde liegenden Wechselwirkungen und von Mediumseffekten abhängig, wobei letztere enthalpischer oder entropischer Natur sein können.
Vereinfachte Betrachtungen:
- Der enthalpische Beitrag zur Komplexbildung spiegelt die Stärke der direkten Wechselwirkungen zwischen Rezeptor und Substrat wider. Die Komplexbildungsenthalpie sollte möglichst negativ sein. Eine positive Komplexbildungsenthalpie kann aber durch eine stark positive Komplexbildungsentropie überkompensiert werden. Die Stärke der Wechselwirkung ist sehr empfindlich von Mediumseffekten (Art und Polarität des umgebenden Lösungsmittels bzw. der Rezeptorkavität) abhängig.
- In erster Näherung sollte die Komplexbildungentropie ungünstig, also negativ sein, da bei der Komplexbildung aus zwei Molekülen ein Komplex wird.
- Bei der Komplexbildung werden Konformationsfreiheitsgrade des Rezeptors und des Substrats eingeschränkt (Flexibilität geht verloren, was entropisch ebenfalls ungünstig ist).
Wie kann eine Komplexbildung dann jemals entropisch günstig werden?
Jede Komplexbildung findet in einem Lösungsmittel statt. Die Einflüsse von Lösungsmittel auf Komplexbildungsgleichgewichte dürfen nicht vernachlässigt werden.
Lösungsmittel können die Komplexbildung enthalpisch beeinflussen. Sind z.B. Rezeptorkavität oder Substrat gut solvatisiert, kostet es Energie, die Solvathülle abzustreifen. Diese Energie wird durch die Wechselwirkungsenergie der Bindungspartner aufgebracht. Die Wechselwirkungsenergie reduziert sich also um diesen Betrag und ein Komplex ist instabiler als in Lösungsmitteln, in denen die Bindungspartner nur schwach solvatisiert werden.
Fazit: In unpolaren Lösungsmitteln ist die Wechselwirkung üblicherweise stärker als in polaren!
Bei der Komplexbildung kommt es zudem zu einer Reorganisation von Lösungsmittelmolekülen. Diese kann erheblich sein und führt in der Regel zu einem Zustand größerer Unordnung. Eine positive Komplexbildungsentropie ist also oft (allerdings nicht notwendigerweise) auf die Freisetzung von Lösungsmittelmolekülen aus den Solvathüllen von Rezeptor und/oder Substrat zurückzuführen. Dieser entropische Beitrag kann eine positive Komplexbildungsenthalpie überkompensieren und die Komplexbildung insgesamt zu einem exergonischen Prozess machen.
Wir werden auf dieses Phänomen u.a. bei der Besprechung von hydrophoben Wechselwirkungen zurückkommen. Die freie Energie der Komplexbildung setzt sich also aus mehreren, wenigstens drei Energiebeiträgen zusammen.
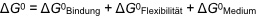
Kinetik
Kinetische Aspekte einer Komplexbildung
Die Komplexbildung ist üblicherweise schnell, d.h. mit einer geringen Aktivierungsenergie verbunden.
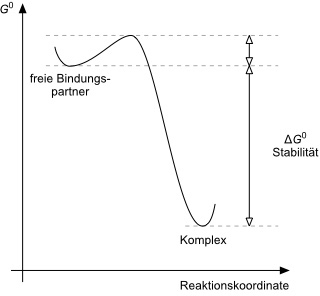
Schnelle Komplexbildung / Hohe thermodynamische Stabilität
Es gibt aber Ausnahmen. Sogenannte Carceranden (siehe Resorcinarene) bilden kinetisch inerte Komplexe, z.B. mit Dimethylacetamid, die aber thermodynamisch nicht besonders stabil sind. Die langsame Dissoziation eines Komplexes bedeutet also nicht zwingend eine hohe thermodynamische Stabilität.
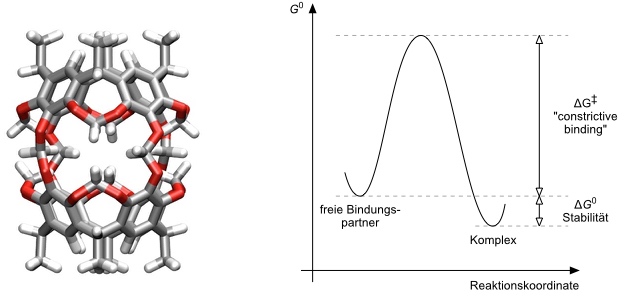
Langsame Komplexbildung / Niedrige thermodynamische Stabilität
Zusammenhang zwischen Kinetik und Thermodynamik
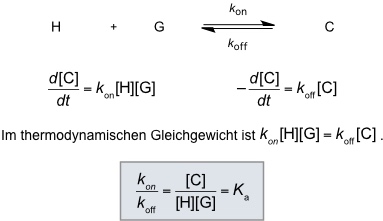
Die thermodynamische Stabilität eines Komplexes ergibt sich also aus dem relativen Verhältnis der Geschwindigkeitskonstanten seiner Bildung bzw. Dissoziation. Bei Carcerandkomplexen ist die Dissoziation langsam aber die Bildung nur wenig schneller. Besonders stabile Komplexe sind zu erwarten, wenn die Bildung schnell ist aber die Dissoziation um ein Vielfaches langsamer (unter Standardbedingungen).
Beispiel:
- Für eine diffusionskontrollierte Reaktion beträgt kon ca. 109 M-1s-1.
- Beträgt die Lebensdauer τ des Komplexes 1 s ist koff = 1 s-1 (τ = 1/koff).
- Es ergibt sich also ein Ka von 109 M-1.
- Typischerweise sind Komplexe in der supramolekularen Chemie deutlich weniger stabil, haben also auch eine erheblich geringere Lebensdauer.
Elektrostatische WW
Entgegengesetzte Ladungen ziehen sich an. Dieser Stärke dieser Wechselwirkungen wird mathematisch mit Hilfe des Coulomb-Gesetzes beschrieben.
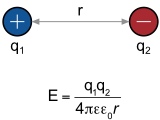
| q1, q2 | Dielektrizitätskonstante des Mediums |
| ε0 | Dielektrizitätskonstante des Vakuums (8,854 · 10-12 C V-1 m-1) |
| r | Abstand der Ionen |
Die Abhängigkeit von der Dielektrizitätskonstante des Mediums führt dazu, dass elektrostatische Wechselwirkungen mit zunehmender Polarität des Mediums schwächer werden.
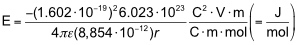
| q1, q2 | 1,602 · 10-19 C |
| ε | 1 (Vakuum), 4,8 (Chloroform), 78 (Wasser) |
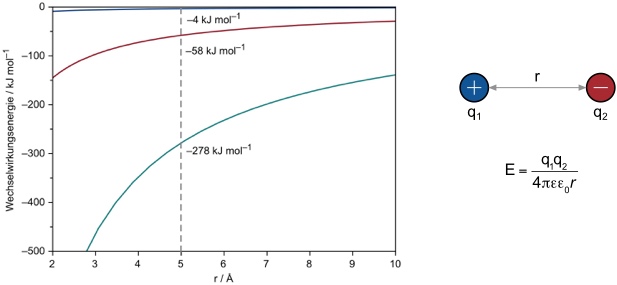
Die Graphik zeigt, dass Ion-Ion-Wechselwirkungen im besten Fall Wechselwirkungsenergien von 100 - 350 kJ mol-1 erreichen können.
Ion-Ion WW
Auf atomarer Ebene ist das Coulomb Gesetz auf die Wechselwirkung von Ionen anwendbar. Das Gesetz gilt streng allerdings nur für Punktladungen. Die Wechselwirkung größerer organische Ionen wird nur bedingt beschrieben. Sie ist in der Regel deutlich schwächer als durch das Coulomb Gesetz vorausgesagt, da die organischen Reste die Ladung der Ionen abschirmen.
Darüber hinaus werden in organischen Verbindungen Ladungen über weite Teile der Moleküle verteilt. So zeigen Berechnungen, dass die positive Ladung im Tetramethylammoniumkation keineswegs auf dem Stickstoffatom lokalisiert ist, wie es die Lewis-Struktur suggeriert. Aufgrund der höheren Elektronegativität von Stickstoff befindet sich die positive Ladung auch auf den Methylgruppen, wie die elektrostatische Potentialoberfläche des Tetramethylammoniumions zeigt.

Außerdem wird durch das Coulomb Gesetz der Einfluss des Lösungsmittels durch Berücksichtigung der Dielektrizitätskonstante nur dann richtig beschrieben, wenn die Lösungsmittelmoleküle keine spezifischen Wechselwirkungen mit den gelösten Ionen eingehen.
Beispiel: Hydratation von Kationen (links) und Anionen (rechts) in Wasser.
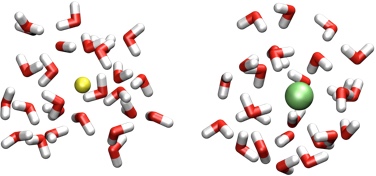
Solche Solvatationseffekte können (insbesondere in Wasser) dazu führen, dass die Wechselwirkung von Kationen und Anionen endotherm also enthalpisch ungünstig ist, da für die Zusammenlagerung der Ionen Wasserstoffbrücken und Wechselwirkungen der Wassermoleküle mit den gelösten Ionen aufgebrochen werden müssen. Allenfalls entropische Effekte können dann die Wechselwirkung begünstigen.
In Proteinen findet die Bindung häufig in einem gut vom umgebenden wässrigen Medium abgeschirmten Hohlraum statt, dessen Dielektrizitätskonstante klein ist und in dem Wechselwirkungen mit Wassermolekülen ausgeschlossen sind. Dies führt zu einer Verstärkung der Bindung im Vergleich zu derselben Wechselwirkung in Wasser.
Bis auf wenige Ausnahmen sind die Wechselwirkungsarten in supramolekularen Systemen alle elektrostatischer Natur, beruhen also auf der Anziehung gegensätzlicher Ladungen oder Partialladungen.
Ion-Dipol WW
Die Wechselwirkungen zwischen Ionen und Molekülen, die aufgrund von polarisierten Bindungen ein Dipolmoment besitzen, wird durch folgende Beziehung beschrieben.
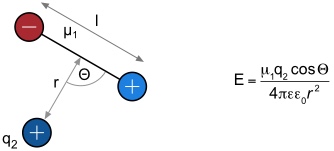
| µ | Dipolmoment des beteiligten Moleküls |
| Θ | Winkel der Punktladung zum Dipolmoment |
Ion-Dipol-Wechselwirkungen treten zwischen Alkalimetallkationen und Wassermolekülen in wässrigen Lösungen auf und sind für die Hydratation von diesen Ionen verantwortlich. Außerdem stabilisieren Ion-Dipol-Wechselwirkungen die Komplexe zwischen Alkalimetallkationen und Kronenethern.
Hydratationsenthalpien einfacher sphärischer Kationen und Anionen geben Informationen zur Abhängigkeit der Stärke von Ion-Dipol-Wechselwirkungen von der Art und den Eigenschaften des beteiligten Ions.
| Kation | Hydratationsenthalpie ΔH0 | Ionenradius | Anion | Hydratationsenthalpie ΔH0 | Ionenradius |
| kJ mol-1 | Å | kJ mol-1 | Å | ||
| Li+ | -510 | 0,6 | |||
| Na+ | -410 | 0,95 | F- | -477 | 1,36 |
| K+ | -339 | 1,33 | Cl- | -343 | 1,81 |
| Rb+ | -318 | 1,48 | Br- | -330 | 1,96 |
| Cs+ | -297 | 1,69 | I- | -272 | 2,16 |
| Mg2+ | -1992 | 0,65 | |||
| Ca2+ | -1661 | 0,90 |
- Die Hydratationsenthalpie von Ionen gleicher Ladung ist umso stärker negativ, je kleiner das Ion ist. Dies ist auf die höhere Ladungsdichte kleinerer Ionen zurückzuführen.
- Einfach geladene Anionen haben einen größeren Ionenradius als die entsprechenden isoelektronischen Kationen (Na+/F-; K+/Cl-), gleichzeitig ist die Hydratationsenthalpie stärker negativ.
- Bei vergleichbaren Ionenradien besitzen Ionen mit höherer Ladung eine stärker negative Hydratationsenthalpie.
Dipol-Dipol WW
Diese Wechselwirkungsart tritt zwischen Molekülen auf, welche aufgrund von polarisierten Bindungen Dipolmomente besitzen.
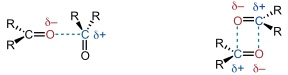
Folgende Beziehung stellt einen Zusammenhang zwischen der Stärke der Wechselwirkungen und den Dipolmomenten der Bindungspartner sowie der Geometrie der Anordnung her.
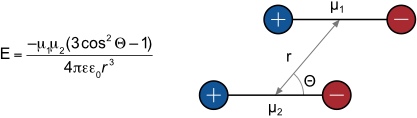
Diese Beziehung gilt nur für parallel angeordnete Dipole. In diesem Fall wird die Wechselwirkung Null wenn Θ 54,7° beträgt. Sind die Dipole in einem Winkel zueinander angeordnet, wird die Beziehung komplexer. Zu beachten ist fernerhin die Abstandsabhängigkeit mit r-3.
Wasserstoffbrücken
Wasserstoffbrücken sind ein Sonderfall von Dipol-Dipol Wechselwirkungen, bei denen ein Wasserstoffatom, das an ein elektronegatives Atom gebunden ist, mit dem negativen Ende eines zweiten Dipols (Atom, funktionelle Gruppe, π-System) wechselwirkt.
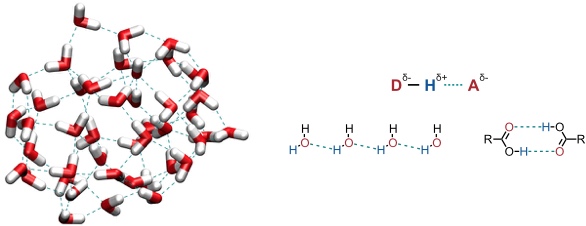
Da Wasserstoffbrücken im Wesentlichen auf elektrostatischen Wechselwirkungen beruhen, hat die Direktionalität der Wechselwirkung nur einen untergeordneten Effekt auf die Bindungsstärke. Dennoch sind lineare Wasserstoffbrücken häufig stabiler als gewinkelte. Darüber hinaus beobachtet man in Systemen mit freien Elektronenpaaren, dass die Bindungspartner bevorzugt Geometrien annehmen, in denen sich die Wasserstoffbrücke in der Achse des freien Elektronenpaars befindet.
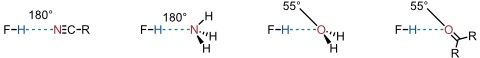
Wichtige Einflüsse auf die Stärke von Wasserstoffbrücken hat die Polarität des Lösungsmittels und die Fähigkeit des Lösungsmittels, selber als Wasserstoffbrückenakzeptor oder -donor zu fungieren.

Dies ist der Grund, warum das Dimer von N-Methylacetamid in Tetrachlormethan nachweisbar ist, in 1,4-Dioxan aber nicht, obwohl beide Lösungsmittel etwa dieselbe Dielektritätskonstante besitzen. Der Grund ist, dass 1,4-Dioxan ist ein Wasserstoffbrückenakzeptor ist und Tetrachlormethan nicht.
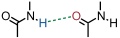
Wasserstoffbrücken sind um so stärker, je höher die positive Partialladung auf dem Wasserstoffatom ist, d.h. je elektronegativer D ist. Wasserstoffbrücken zu den Protonen von Halogenwasserstoffsäuren werden daher in folgender Reihenfolge zunehmend schwächer:
HF > HCl > HBr > HI
Die Abhängigkeit der Stärke einer Wasserstoffbrücke von der Elektronegativität von A ist komplexer, da elektronegative Heteroatome trotz hoher negativer Partialladung wenig geneigt sind, ihre Elektronendichte mit Bindungspartnern zu teilen. Wasserstoffbrücken zu Fluor sind daher schwach. Auch Wasserstoffbrücken zu Heteroatomen höherer Periode sind aufgrund der diffusen Elektronenhüllen schwach. Wasserstoffbrücken zu den Heteroatomen in der folgenden Verbindungen werden daher zunehmend schwächer:
H2O > H3N > H2S > H3P
Weitere Effekte, die die Stärke einer Wasserstoffbrücke beeinflussen sind Resonanz-Verstärkung und Polarisations-Verstärkung.
Resonanz-Verstärkung
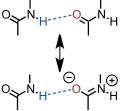
Polarisations-Verstärkung.

Durch Anlagerung des linken Wassermoleküls verändert sich die negative Partialladung auf dem mittleren Sauerstoffatom über die H–O Bindung. Dadurch wird die Bindung zum an das in die Wasserstoffbrücke involvierte Proton des rechten Wassermoleküls verstärkt.
Unter sekundären Wasserstoffbrücken versteht man die attraktive und repulsive Wechselwirkungen zwischen benachbarten Wasserstoffbrückenakzeptoren und -donoren.
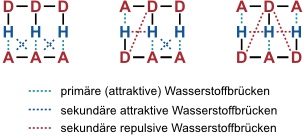
Diese Wechselwirkungen erklären beispielsweise, warum das Cytosin-Guanin-Dimer stabiler ist als das Uracil-2,6-Diaminopyridin-Dimer.
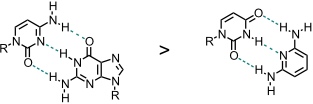
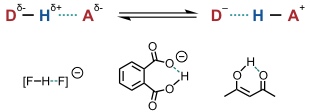
Sogenannte low-barrier-Wasserstoffbrücken findet man in Systemen, in denen die vollständige Protonenübertragung vom Wasserstoffbrückendonor auf den -akzeptor zu einem thermodynamisch gleich stabilen System führt und die Aktivierungsenergie für die Protonenübertragung gering ist. Diese Wasserstoffbrücken können sehr stabil sein, sind aber auf Sonderfälle begrenzt.
Kation-π WW
Unter Kation-π Wechselwirkungen versteht man elektrostatische Wechselwirkungen zwischen Kationen und den negativen Potentialoberflächen von π-Systemen. Diese Wechselwirkungsart ist mit der Ion-Dipol Wechselwirkung vergleichbar. Allerdings haben Moleküle wie Ethylen oder Benzol kein Dipolmoment, sondern ein permanentes Quadrupolmoment.
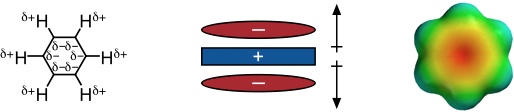
Das elektrostatische Potential von Benzol ist dementsprechend auf den Oberflächen des Ringes negativ und in der Peripherie positiv. Kationen können also mit Flächen von aromatischen Ringsystemen wechselwirken.
Neben Kristallstrukturen von Proteinen mit gebundenen Kationen lieferten Modellverbindungen Hinweise auf die Bedeutung von Kation-π Wechselwirkungen. Wichtige Beiträge in diesem Zusammenhang kamen von der Gruppe um G. W. Gokel. So ist der Aromat in folgendem Kronenether im Kristall über dem gebundenen Natriumion angeordnet, was für eine Wechselwirkung zwischen Gast und π-System spricht.
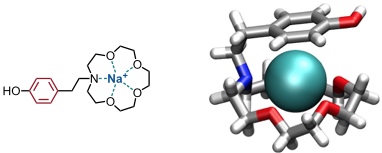
[Für eine interaktive Version der Kristallstruktur hier klicken]
[Quelle: Hu, J.; Barbour, L. J.; Ferdani, R.; Gokel, G. W. Chem. Commun. 2002, 1810-1811: <doi>.]
In der Gasphase können die Bindungsstärken erheblich sein. Die folgende Tabelle zeigt die Stärke der Wechselwirkung von Alkalimetallkationen mit Benzol in der Gasphase.
| M+ | ΔG0 / kJ mol-1 |
| Li+ | 159 |
| Na+ | 113 |
| K+ | 79 |
| Rb+ | 67 |
[Quelle: E. V. Anslyn, D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, CA, 2004, S. 181.]
Die Wechselwirkung ist um so stärker, je höher die Ladungsdichte des Kations ist. Es ist zu beachten, dass Kation-π Wechselwirkungen beim Übergang von der Gasphase in protische Lösungsmittel aufgrund von hydrophoben Wechselwirkungen weniger stark geschwächt werden als Ion-Dipol Wechselwirkungen.
Die Stärke von Kation-π Wechselwirkungen wird durch Substituenteneffekte beeinflusst. Dabei sind induktive Effekte ebenso wichtig wie mesomere.
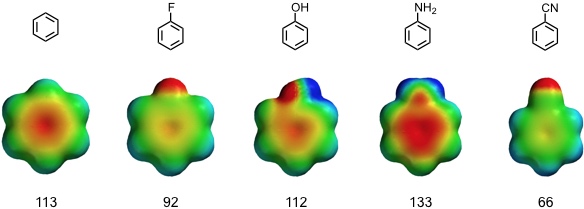
Berechnete Bindungsenergien von Na+ in kJ mol-1
In Analogie zu Dipol-Dipol Wechselwirkungen können auch Moleküle mit polarisierten Bindungen mit den negativen Potentialoberflächen ungesättigter Verbindungen wechselwirken. Diese Art von Wechselwirkung wird Polar-π- Wechselwirkung genannt. Sie ist jedoch schwach.
Literatur
- Ma, J. C.; Dougherty, D. A. "The Cation-π Interaction" Chem. Rev. 1997, 97, 1303-1324: <doi>.
- Gokel, G. W.; De Wall, S. L.; Meadows, E. E. "Experimental Evidence for Alkali Metal Cation-π Interactions" Eur. J. Org. Chem. 2000, 2967-2978: <doi>.
- Gokel, G. W. "The Aromatic Sidechains of Amino Acids as Neutral Donor Groups for Alkali Metal Cations" Chem. Commun. 2003, 2847-2852: <doi>.
- Dougherty, D. A. "The Cation-π Interaction" Acc. Chem. Res. 2013, 46, 885-893: <doi>.
Anion-π WW
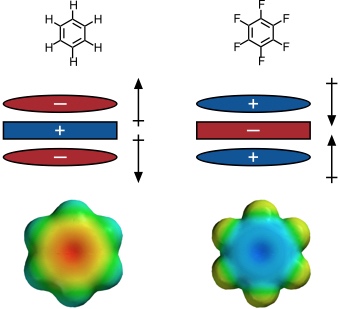
Anion-π Wechselwirkungen treten zwischen elektronenarmen π-Systemen und Anionen auf.
Elektrostatische Wechselwirkungen mit den Quadrupolmomenten aromatischer Ringe.
Elektrostatische Wechselwirkungen basierend auf Anion-induzierten Dipolmomenten.
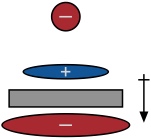
Um starke Anion-π Wechselwirkungen zu ermöglichen, muss ein aromatischer Ring also ein großes positives Quadrupolmoment besitzen und gut polarisierbar sein.
Informationen über die Existenz und die potentielle Stärke von Anion-π Wechselwirkungen lieferten insbesondere theoretischen Berechnungen und Kristallstrukturen. Beispiele für synthetische Rezeptoren, in denen Anion-π Wechselwirkungen zur Substratbindung beitragen, sind selten.
Beispiel 1
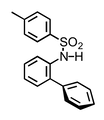 | 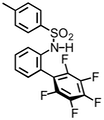 | |
| Anion | Ka / M-1 | Ka / M-1 |
| Cl- | <1 | 30 |
| Br- | <1 | 20 |
| I- | <1 | 34 |
Lösungsmittel: CDCl3.
In diesem Fall muss berücksichtigt werden, dass die elektronischen Eigenschaften des aromatischen Ringes auch die Acidität der NH Gruppe beeinflussen.
[Quelle: Berryman, O. B.; Hof, F.; Hynes, M. J.; Johnson, D. W. Chem. Commun. 2006, 506-508: <doi>.]
Beispiel 2
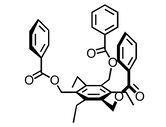 | 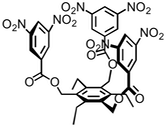 | |
| Anion | Ka / M-1 | Ka / M-1 |
| Cl- | <1 | 53 |
| Br- | <1 | 35 |
| I- | <1 | 26 |
Lösungsmittel: CDCl3.
Strukturelle Untersuchungen weisen darauf hin, dass in diesem Fall andere Bindungsmodi (σ···X– oder C–H···X– Wechselwirkungen) an der Anionenbindung der Derivate mit elektronenziehenden Substituenten beteiligt sind.
[Quelle: Berryman, O. B.; Sather, A. C.; Hay, B. P.; Meisner, J. S.; Johnson D. W. J. Am. Chem. Soc. 2008, 130, 10895-10897: <doi>.]
Wegen der im Vergleich zu Kationen größeren Ionenradien von Anionen sind Anion-π Wechselwirkungen schwächer als Kation-π Wechselwirkungen.
Literatur
- Schottel, B. L.; Chifotides, H. T.; Dunbar, K. R. "Anion-π Interactions" Chem. Soc. Rev. 2008, 37, 68-83: <doi>.
- Frontera, A.; Gamez, P.; Mascal, M.; Mooibroek, T. J.; Reedijk, J. "Anion-π Wechselwirkungen ins rechte Licht gerückt" Angew. Chem. 2011, 123, 9736-9756: <doi>.
- Giese, M.; Albrecht, M.; Rissanen, K. "Experimental investigation of anion-π interactions - applications and biochemical relevance" Chem. Commun. 2016, 52, 1778-1795.: <doi>.
π-π WW
Aufgrund des Quadrupolmoments können die π-Systeme ungesättigter Verbindungen miteinander wechselwirken. Diese Art der Wechselwirkung wird π-π Wechselwirkung genannt. Sie führt bevorzugt zu einer T-förmigen (edge to face) oder einer versetzten Anordnung zweier (identischer) π-Systeme. Eine exakte Stapelung ist aufgrund von abstoßenden Wechselwirkungen nicht möglich.
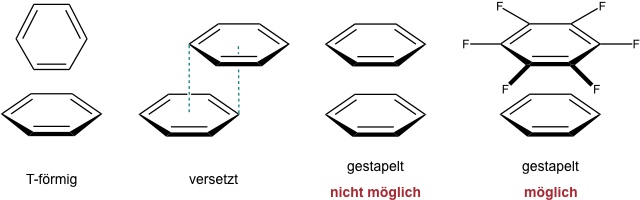
Interessanterweise haben Benzol und Hexafluorbenzol exakt komplementäre Quadrupolmomente. Im Gegensatz zu reinem Benzol oder reinem Hexafluorbenzol ordnet sich eine 1:1 Mischung dieser Verbindungen im Kristall daher in einer stapelförmigen Anordnung an.
Charge-Transfer WW
Eine besondere Form von π-π Wechselwirkungen sind Charge-Transfer Wechselwirkungen. Diese werden zwischen elektronenreichen π-Systemen mit hoch liegendem HOMO und elektronenarmen π-Systemen mit niedrig liegendem LUMO beobachtet. Bei der Wechselwirkung kommt es zu einer stabilisierenden Orbitalwechselwirkung. Es handelt sich in diesem Fall also nicht um elektrostatische Wechselwirkungen.
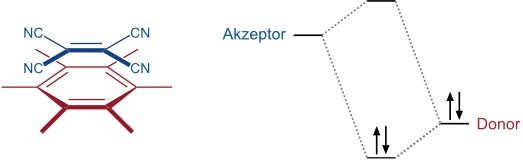
Charge-Transfer Wechselwirkungen äußern sich meist in einem intensiven Übergang im UV/Vis-Spektrum, welcher auf eine elektronische Anregung vom HOMO ins LUMO des Komplexes zurückzuführen ist. Hierbei erfolgt eine Ladungstrennung. Die Stabilität eines Charge-Transfer Komplexes ist allerdings auf die Orbitalwechselwirkung im Grundzustand und nicht auf die Ladungstrennung im angeregten Zustand zurückzuführen.
Charge-Transfer Wechselwirkungen spielen bei den ivon J. F. Stoddart beschriebenen Rotaxanen und Catenanen eine Rolle.
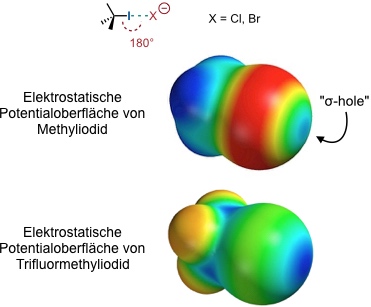
Halogen WW
Unter Halogen Wechselwirkungen versteht man die Wechselwirkung eines Halogenids (meist Chlorid oder Bromid) mit einem elektronenarmen, organischen Iodid oder Bromid. Die Wechselwirkung kann als elektrostatische Wechselwirkung aufgefasst werden. Sie hat eine wesentlich höhere Direktionalität als Wasserstoffbrücken, wobei die optimale Anordnung der Bindungspartner eine lineare ist.
Der folgende Rezeptor zeigt den Einfluss von Halogen Wechselwirkungen auf die Anionenaffinität.
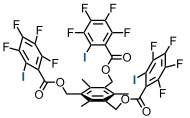
| Anion | Ka / M-1 |
| Cl- | 19000 |
| Br- | 3800 |
| I- | 760 |
| TsO- | 10 |
| HSO4- | <10 |
| NO3- | <10 |
Lösungsmittel: Aceton; T = 295 K; Gegenionen: (nBu)4N+.
[Quelle: Sarwar, M. G.; Dragisic, B.; Sagoo, S.; Taylor, M. S. Angew. Chem. 2010, 122, 1718-1721: <doi>.]
Literatur
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. "Halogen bonding in supramolecular chemistry" Angew. Chem. 2008, 120, 6206-6220: <doi>.
- Cavallo, G.; Merangolo, P.; Milani, R.; Pilati, T.; Triimagi, A.; Resnati, G.; Terraneo, G. "The Halogen Bond" Chem. Rev. 2016, 116, 2478-2601: <doi>.
van-der-Waals WW
Unter van-der-Waals Wechselwirkungen oder London-Dispersions Wechselwirkungen versteht man Wechselwirkungen zwischen den induzierten Dipolen zweier polarisierbarer Moleküle. Die Abstandsabhängigkeit wird mathematisch durch das Lennard-Jones Potential beschrieben.
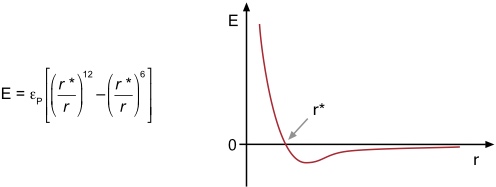
Für r > r* wird E geringfügig negativ also attraktiv, während für r < r* eine repulsive Wechselwirkung resultiert, die durch die Abstoßung der Elektronenhüllen zweier Moleküle zu erklären ist, wenn diese sich zu nahe kommen. Der Term εP beschreibt die Polarisierbarkeit der Elektronensysteme der beteiligten Bindungspartner. Bei Wechselwirkung zweier besser polarisierbarer Systeme ist der Anstieg der repulsiven Wechselwirkungen etwas weniger steil als bei Wechselwirkung zweier weniger gut polarisierbarer Verbindungen.
Weitere elektrostatische Wechselwirkungen:
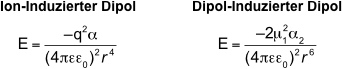
Hydrophobe WW
Diese Wechselwirkungen sind im Wesentlichen auf Lösungsmitteleffekte zurückzuführen, welche zudem weitgehend auf das Lösungsmittel Wasser beschränkt sind.
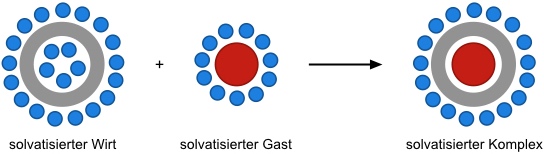
Die Solvatation kleiner unpolarer Moleküle in Wasser ist meist nur geringfügig endotherm oder sogar enthalpisch neutral. Allerdings ist sie durch die Strukturierung der Wassermoleküle um das organische Molekül entropisch stark benachteiligt. Lagern sich zwei oder mehrere organische Moleküle in Wasser daher zusammen (Phasentrennung), ist dies mit einem entropischen Gewinn des Gesamtsystems verbunden. Hydrophobe Wechselwirkungen sind daher in den vielen Fällen entropiegetrieben. Im übertragenen Sinn gelten diese Betrachtungen auch für die Bildung von Wirt- Gast Komplexen in Wasser.
Bei größeren organischen Molekülen kann eine Solvatation in Wasser enthalpisch ungünstig sein. Eine Aggregation ist in diesem Fall sowohl enthalpisch als auch entropisch günstig.
Literatur
- Smithrud, D. B.; Sanford, E. M.; Chao, I.; Ferguson, S. B.; Carcanague, D. R.; Evansek, J. D.; Houk, K. N.; Diederich, F. "Solvent effects in molecular recognition" Pure Appl. Chem. 1990, 62, 2227-2236: <doi>.
Quantifizierung
Massenwirkungsgesetz
Bei der Charakterisierung von Komplexen aus synthetischen Rezeptoren mit den entsprechenden Substraten ist man vorrangig an der Komplexstabilität interessiert. Diese wird quantitativ durch das Massenwirkungsgesetz beschrieben.
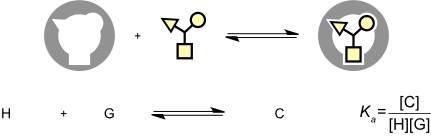
Dieser Zusammenhang gilt aber nur für 1:1 Komplexe. Bei Komplexen höherer Stöchiometrie werden die Zusammenhänge etwas komplizierter. Aus diesem Grund ist es unerlässlich, vor der Bestimmung der Stabilitätskonstante eines Komplexes zunächst dessen Zusammensetzung zu untersuchen.
Weiterhin muss berücksichtigt werden, dass im Massenwirkungsgesetz Konzentrationen der Bindungspartner und des Produktes im thermodynamischen Gleichgewicht auftreten. Diese sind von den eingesetzten Konzentrationen der Bindungspartner verschieden und für die Quantifizierung einer Stabilitätskonstante muss man dementsprechend Methoden anwenden, die die Bestimmung der im Massenwirkungsgesetz auftretenden Konzentrationen gestatten.
Bestimmung der Komplexstöchiometrie - Methode der kontinuierlichen Variationen - Job Plot
Dieses Verfahren wird häufig zur Bestimmung der Stöchiometrie eines Komplexes eingesetzt. Das zugrundeliegende Prinzip kann man qualitativ leicht verstehen. In allgemeiner Form kann man das Gleichgewicht der Bildung eines beliebigen Komplexes folgendermaßen formulieren.
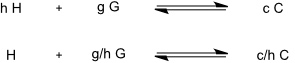
Für einen Job Plot wird eine Serie von Lösungen hergestellt, deren Gesamtkonzentrationen c0 = [H]0 + [G]0 jeweils identisch sind. Das Verhältnis der Bindungspartner (bzw. der Molenbruch X = [H]0 / ([H]0 + [G]0) wird jedoch über die Serie von Lösungen variiert. Anschließend wird eine Messung durchgeführt, die Informationen über die Menge des gebildeten Komplexes in Lösung liefert. Unter diesen Bedingungen ist die Komplexkonzentration innerhalb dieser Serie in derjenigen Probe am größten, in der das stöchiometrische Verhältnis der Bindungspartner gerade X = h/(h+g) entspricht.
Job, P. Ann. Chim. Appl. 1928, 9,113-203.
Mathematisch ergeben sich für das Beispiel eines 1:1 Komplexes folgende Zusammenhänge.

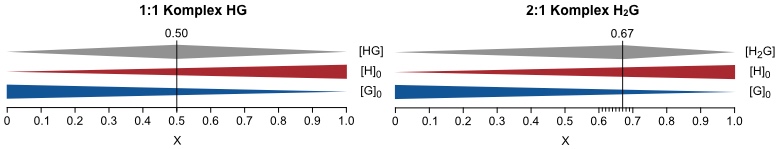
In einem Job-Plot wird das Produkt des Molenbruchs X mit einer Messgröße, die proportional zu [C] ist, gegen den Molenbruch des Rezeptors X aufgetragen. Die Abbildung zeigt Job Plots für 1:1 Komplexe HG unterschiedlicher Stabilität.
Die Abbildung zeigt, dass der Kurvenverlauf bei einem Job-Plot einer Glockenkurve entspricht. Dabei befindet sich das Maximum der Kurve bei 1:1 Komplexen bei X = 0.5. Die Form der Kurve hängt außerdem von den Ausgangskonzentrationen [H]0 und [G]0 und von der Stabilitätskonstante des Komplexes ab.
Analoge Betrachtungen können für Komplexe mit höherer Stöchiometrie durchgeführt werden. So erhält man für 2:1 Komplexe ebenfalls Glockenkurven mit einem Maximum bei X = 0.33 oder X = 0.67, je nachdem, ob der Komplex zwei Rezeptor oder zwei Gastmoleküle enthält.
Bestimmung der Stabilitätskonstante von 1:1 Komplexen - Wirt-Gast Titrationen
Die Bestimmung von Stabilitätskonstanten erfolgt mittels sogenannter Wirt-Gast Titrationen. Dabei wird wiederum eine Serie von Lösungen hergestellt, die unterschiedliche Ausgangskonzentrationen von Wirt [H]0 und Gast [G]0 enthalten. Häufig wird eine dieser Konzentrationen konstant gehalten und die andere variiert. Dann wird mit einem geeigneten Messverfahren die Gleichgewichtslage in den einzelnen Lösungen bestimmt. Dazu wird eine Messgröße bestimmt, die linear mit der Konzentration des Komplexes im Gleichgewicht [C] korreliert.
Die Auftragung dieser Messgröße gegen das Verhältnis [H]0/[G]0 ergibt sogenannte Bindungsisothermen, aus denen die Stabilitätskonstante mittels nicht-linearer Regression ermittelt werden kann. Den zugrundeliegenden mathematischen Formalismus für einen 1:1 Komplex haben wir schon abgeleitet.
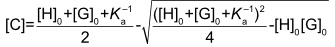
Mit einem Computerprogramm wird Ka so lange variiert bis die gemessenen Werte für [C] mit den berechneten optimal übereinstimmen.
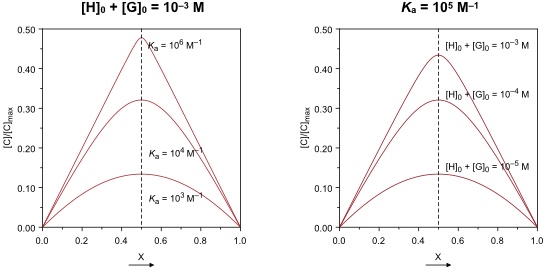
Die folgenden Abbildungen zeigen, dass die Form von Bindungsisothermen von der Stabilitätskonstante des Komplexes und von den Konzentrationen der eingesetzten Bindungspartnern abhängt.
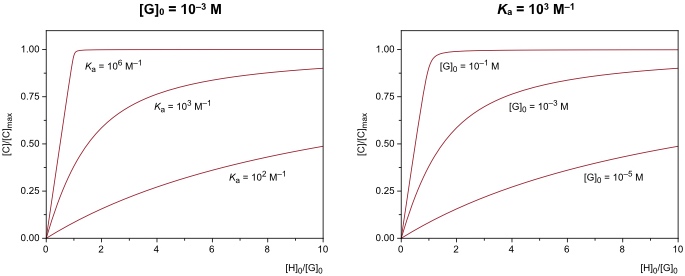
Besonders genaue Ergebnisse werden erhalten, wenn am Ende der Titration der Komplex zu ca. 80 % vorliegt ([C] = 0.8 · [C]max). Erreicht die Bindungsisotherme zu schnell das Plateau oder ist am Ende der Titration der Komplex nur in geringen Mengen gebildet, wird die nicht-lineare Regression ungenau.
Es soll angemerkt werden, dass der Krümmungspunkt der Kurve auch Aussagen über die Komplexstöchiometrie liefert.
Messmethoden
NMR Spektroskopie - Gleichgewichtseinstellung langsam auf der NMR Zeitskala
Komplexierung von Na2SO4 durch ein dreifach verbrücktes Bis(cyclopeptid) in D2O/CD3OD 1:1 (v/v).
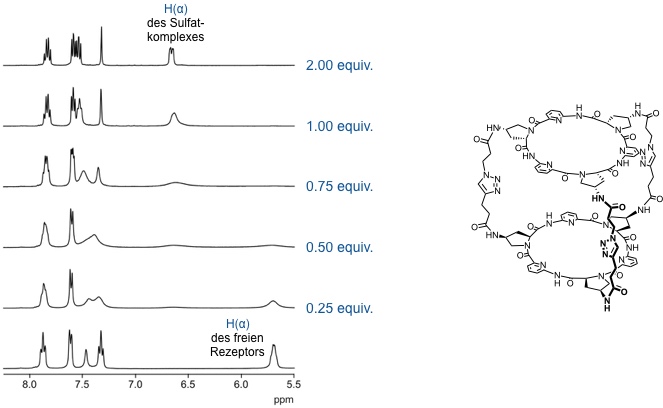
Aus den Integralen äquivalenter Signale des Rezeptors und seines Komplexes im NMR Spektrum kann man das Verhältnis beider Verbindungen im Gleichgewicht direkt ermitteln. Wenn man für die Auswertung keine äquivalenten Signale verwendet, muss die Anzahl der jeweils absorbierenden Protonen mit berücksichtigt werden.
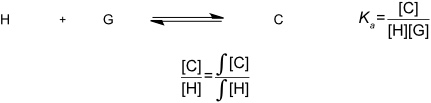
Die jeweiligen Konzentrationen von [C], [H] und [G] ergeben sich aus den Massenbilanzen.

Damit sind alle im Massenwirkungsgesetz auftretenden Konzentrationen bekannt und die Gleichgewichtskonstante kann direkt berechnet werden.
Man bestimmt Ka in der Regel für eine Serie von Spektren und ermittelt dann den Mittelwert, um den Fehler zu minimieren.
NMR Spektroskopie - Gleichgewichtseinstellung schnell auf der NMR Zeitskala
Komplexierung von Natriumtosylat durch ein Cyclopeptid in d6-DMSO.
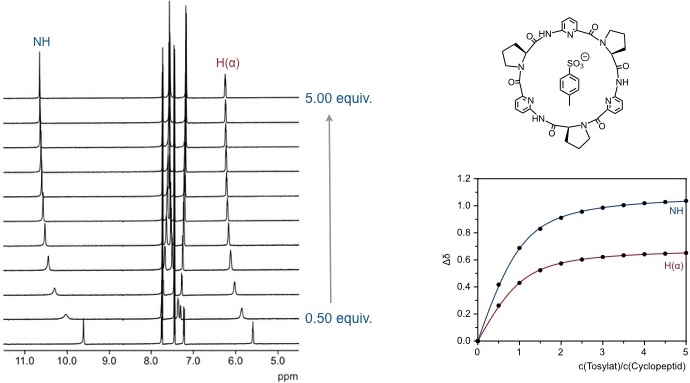
In diesem Fall ist das Ausmaß der Verschiebung des beobachteten Signals proportional zur Konzentration des gebildeten Komplexes. Der mathematische Zusammenhang lautet folgendermaßen.
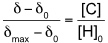
Hierin ist δ die beobachtete chemische Verschiebung des Signals, δ0 die Verschiebung des Signals zu Beginn der Titration und δmax die Verschiebung bei vollständiger Komplexbildung. Dieser Zusammenhang gilt, wenn man ein Signal des Rezeptors verfolgt. Ansonsten steht im Nenner des rechten Terms [G]0.
Einsetzen der bereits abgeleiteten Formel für die Berechnung von [C] führt zu folgendem Ausdruck.
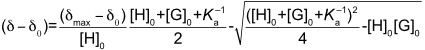
Darin sind Ka und δmax Unbekannte. Mittels eines Computerprogramms und nicht-linearer Regression werden nun diese beiden Unbekannten so lange variiert, bis die berechnete Bindungsisotherme mit der experimentell bestimmten optimal übereinstimmt.
UV/Vis bzw. Fluoreszenz Spektroskopie
Komplexierung von Glucose durch ein fluoreszenzaktives Cyclopeptid mit zwei Boronsäuregruppen.
Im Unterschied zur NMR Spektroskopie, bei der die Lage eines Signals kaum von der Konzentration abhängt, gibt es einen linearen Zusammenhang in der UV/Vis- bzw. Fluoreszenz-Spektroskopie zwischen Signalintensität und Konzentration (Lambert-Beer-Gesetz). Ändert sich also bei einer Komplexbildung die Intensität einer Bande und verändert man während der Wirt-Gast-Titration zudem die Konzentration des dort absorbierenden Bindungspartner, kommt es zu überlagerten Effekten. Man muss daher bei der Auswertung die Extinktionskoeffizienten der jeweiligen Bindungspartner berücksichtigen.
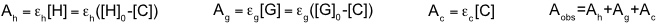
Die Zusammenhänge vereinfachen sich, wenn nicht alle der am Gleichgewicht beteiligten Komponenten im untersuchten Wellenlängenbereich absorbieren bzw. emittieren.
Potentiometrie
Komplexierung von 1,2,3-Benzoltricarbonsäure (1,2,3-BTC) und Citronensäure (CIT) durch ein makrocyclisches Heptaamin ([21]aneN7).
Logarithmen der kumulativen und schrittweisen Protonierungskonstanten
| Reaktion | 1,2,3-BTC | CIT |
| A3- + H+ ⇌ HA2- | 5,501 | 5,441 |
| A3- + 2 H+ ⇌ H2A- | 9,300 | 9,615 |
| A3- + 3 H+ ⇌ H3A | 11,945 | 12,359 |
| HA2- + H+ ⇌ H2A- | 3,90 | 4,14 |
| H2A- + H+ ⇌ H3A | 2,65 | 2,74 |
Lösungsmittel: 0.15 M wässrige NaClO4, T = 298 K.
Die beiden Carbonsäuren haben analoge pKs-Werte und daher kann man die Gleichgewichtskonstanten ihrer Bindung an den Rezeptor direkt vergleichen. Bei potentiometrischen Titrationen erhält man Informationen über die pH Wert abhängigen Gleichgewichtskonstanten der Wechselwirkung zwischen Wirt und Gast in jeweils unterschiedlichen Protonierungsstufen.
Logarithmen der kumulativen und schrittweisen Protonierungskonstanten
| Reaktion | 1,2,3-BTC | CIT |
| L + A3- + 3 H+ ⇌ LAH3 | 30,89 | |
| L + A3- + 4 H+ ⇌ LAH4+ | 39,24 | 36,82 |
| L + A3- + 5 H+ ⇌ LAH52+ | 46,05 | 42,37 |
| L + A3- + 6 H+ ⇌ LAH63+ | 50,97 | 46,53 |
| L + A3- + 7 H+ ⇌ LAH74+ | 54,60 | 50,08 |
| L + A3- + 8 H+ ⇌ LAH85+ | 57,64 | |
| L + A3- + 9 H+ ⇌ LAH9+ | 60,20 | |
| A3- + H3L3+ ⇌ LAH3 | 3,2 | |
| A3- + H4L4+ ⇌ LAH4+ | 5,2 | 2,7 |
| A3- + H5L5+ ⇌ LAH52+ | 8,2 | 4,6 |
| A3- + H6L6+ ⇌ LAH63+ | 11,0 | 6,6 |
| A3- + H7L7+ ⇌ LAH74+ | 12,7 | 8,1 |
Lösungsmittel: 0.15 M wässrige NaClO4, T = 298 K.
Anschaulicher werden die Unterschiede in der Bindung der beiden Carbonsäuren an den Rezeptor wenn man sie graphisch in Verteilungsdiagrammen darstellt
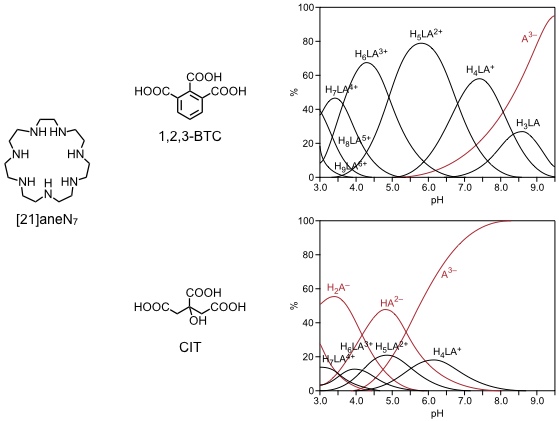
[Quelle: Bencini, A.; Bianchi, A.; Burguete, M. I.; Dapporto, P.; Doménech, A.; García-España, E.; Luis, S. V.; Paoli, P.; Ramírez, L. A. J. Chem. Soc., Perkin Trans. 2 1994, 569-577: <doi>.
Isotherme Titrationskalorimetrie
Messprinzip und Bindungsisotherme
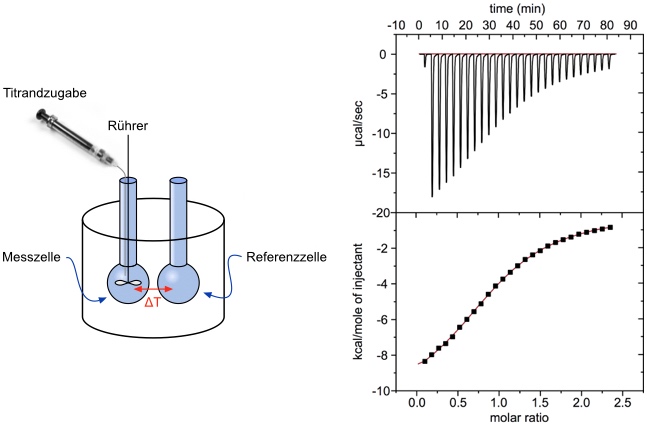
Als Ergebnis der Titration erhält man die Bindungskonstante Ka, den Stöchiometriefaktor n und die Komplexbildungsenthalpie ΔH0. Mit ΔG0 = -RT lnKa und der Gibbs-Helmholtz-Gleichung ΔG0 = ΔH0 - TΔS0 kann man die Komplexbildungsentropie berechnen. Damit sind durch eine einzige Titration alle thermodynamischen Parameter einer Komplexbildung zugänglich. Bei den anderen Verfahren muss man die Stabilitätskonstanten temperaturabhängig bestimmen, um durch eine van't Hoff Auftragung die Reaktionsenthalpie bzw. Reaktionsentropie bestimmen zu können.
Literatur
- Connors, K. A. "Binding constants", Wiley, New York, 1987.
- Fielding, L. "Determination of association constants (Ka) from solution NMR data" Tetrahedron 2000, 56, 6151-6170: <doi>.
- Schalley, C. A. "Analytical Methods in supramolecular chemistry" Wiley-VCH, Weinheim, 2007: <doi>.
- Thordarson, R. "Determining association constants from titration experiments in supramolecular chemistry" Chem. Soc. Rev. 2011, 40, 1305-1323: <doi>.
Letzte Änderung: 23-03-30. Email