Lehre - Spektroskopische Strukturaufklärung organischer Verbindungen - IR-Spektroskopie
Physikalische Grundlagen
Infrarotstrahlung regt in organischen Molekülen Rotationen und Schwingungen an. Dabei absorbieren organische Moleküle typischerweise im Wellenzahlbereich von 4000 bis 400 cm-1. Als vereinfachtes Modell kann die Schwingung der Bindung eines zweiatomigen Moleküls wie die Schwingung einer elastischen Feder zwischen zwei Kugeln der Massen m1 und m2 betrachtet werden.
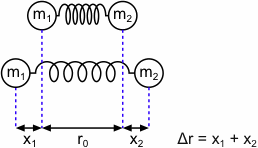
In diesem mechanischen Modell des harmonischen Oszillators ist nach dem Hookschen Gesetz die rücktreibende Kraft F proportional zur Auslenkung Δr und zur Federkonstante k. Die potentielle Energie des harmonischen Oszillators E ist vom Quadrat der Auslenkung abhängig. Somit ergibt sich eine parabelförmige Abhängigkeit der potentiellen Energie von der Auslenkung.
Hooksches Gesetz: F = - k Δr
Potentielle Energie: F = 0,5 k Δr2
Die Berechnung der Energiezustände eines atomaren Systems muss allerdings auf Basis der Schrödingergleichung erfolgen. Eine entsprechende Betrachtung liefert für die Energieeigenwerte eines harmonischen Oszillators folgende Abhängigkeit.
Ev = h νosc (v + 0,5)
h = Plancksches Wirkungsquantum
νosc = Schwingungsfrequenz
v = Schwingungsquantenzahl
Im quantenmechanischen Modell besitzt ein harmonischer Oszillator also diskrete Energieeigenwerte. Die entsprechenden Eigenfunktionen verlaufen innerhalb der Parabel, die durch die Potentialkurve des mechanischen Oszillators begrenzt wird. Mit zunehmender Schwingungsquantenzahl steigt die Anzahl der Knoten in den Eigenfunktionen. Die Energie des Grundzustands mit der Quantenzahl Null bezeichnet man als Nullpunktsenergie. Sie beträgt 0,5 h ν. Nach den Auswahlregeln sind nur Übergänge zwischen benachbarten Energieniveaus erlaubt (Δv = ±1).
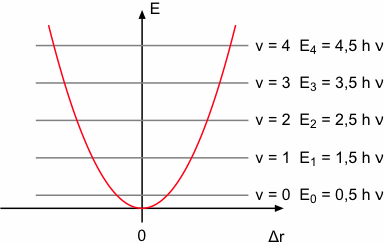
Die Anregung der Schwingung kann man sich anschaulich so vorstellen, dass ein Molekül unter Absorption eines Lichtquants vom Schwingungsgrundzustand in den ersten angeregten Zustand übergeht. Dabei muss die Energie des Lichtquants hν entsprechend der spektroskopischen Resonanzbedingung gerade der Energiedifferenz der beiden Energieniveaus ΔE entsprechen.
Das Modell des harmonischen Oszillators ist jedoch vereinfacht, denn es vernachlässigt die Bindungsdissoziation bei Anregung in hohe Schwingungszustände. Ein realistischeres Bild liefert das Modell des anharmonischen Oszillators. Die Potentialkurve (Morse-Potential) eines anharmonischen Oszillators hat einen asymmetrischen Verlauf, wobei die Kurve bei großen Kernabständen ein Plateau erreicht. Die Energiedifferenz vom Schwingungsgrundzustand bis zu diesem Niveau entspricht der Dissoziationsenergie.
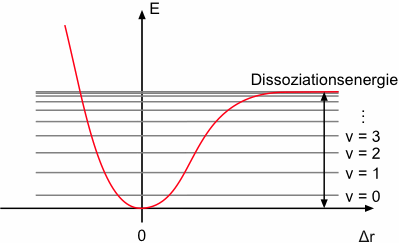
Bei einem anharmonischen Oszillator sind auch Übergänge zwischen nicht benachbarten Schwingungsniveaus erlaubt. Dies führt zu einer Zunahme der Anzahl der Linien in einem Spektrum (Oberschwingungen). Da die wichtigsten Übergänge in der IR-Spektroskopie allerdings zwischen E0 und E1 erfolgen und sich die Potentialkurven eines harmonischen und eines anharmonischen Oszillators in ihren Minima ähneln, werden die meisten Schwingungen in der IR-Spektroskopie näherungsweise mit dem Modell des harmonischen Oszillators beschrieben.
Die Oszillatorfrequenz einer schwingenden Bindung νosc hängt von der Stärke der Bindung, d.h. von der Kraftkonstante k, und von der reduzierten Masse μ gemäß folgender Beziehung ab.
νosc = (2 π)-1 (k / μ)½
Die Schwingungsfrequenz ist danach um so höher, je größer die Kraftkonstante ist, d.h. je stärker die Bindung ist. Umgekehrt bedeutet dies, dass die Anregungsfrequenz der eingestrahlten elektromagnetischen Strahlung um so höher sein muss, je stärker eine Bindung ist. Die zweite Größe in obiger Gleichung ist die reduzierte Masse μ. Sie ist definiert als:
μ = m1 · m2 / (m1 + m2).
Zum einen lässt diese Beziehung erkennen, dass die Frequenz (bzw. die Anregungsenergie) der Schwingung eines zweiatomigen Moleküls um so größer ist, je kleiner die reduzierte Masse der beteiligten Atome. Dabei wird die reduzierte Masse größer wenn die an einer Bindung beteiligten Atome schwerer werden. Zweitens beeinflussen schwere Atome die Schwingungsfrequenz weniger stark als leichte. Letztlich ist dies der Grund, dass einzelne Banden in dem IR-Spektrum eines vielatomigen Moleküls abgekoppelt von den Schwingungen des Restmoleküls den Schwingungen einer einzelnen Bindung zugeordnet werden können.
Neben den quantenmechanischen Auswahlregeln muss man zusätzlich berücksichtigen, dass Schwingungen nur dann IR-aktiv sind, wenn sie eine Änderung des Dipolmoments des Moleküls hervorrufen. Ursache hierfür ist, dass elektromagnetische Strahlung nur dann absorbiert werden kann, wenn das Dipolmoment eines Moleküls mit dem elektrischen Vektor der elektromagnetischen Strahlung in Wechselwirkung treten kann. Als wichtigste Konsequenz dieser Auswahlregel folgt, dass in einem Molekül mit Symmetriezentrumalle Schwingungen, die symmetrisch zum Symmetriezentrum erfolgen, verboten sind.
Die Anzahl der Normalschwingungen eines Moleküls mit n Atomen ergibt sich aus folgenden Beziehungen:
3n - 5 für lineare Moleküle,
3n - 6 für nicht-lineare Moleküle.
CO2 kann demnach vier Normalschwingungen ausführen und CH4 neun. Allerdings muss man berücksichtigen, daß manche Schwingungsmoden entartet und andere IR-inaktiv sein können, was zu einer Verringerung der Zahl der Banden im IR-Spektrum führt. So ist die symmetrische Streckschwingung im CO2 IR-verboten und die beiden Beugeschwingungen sind entartet, so dass im Spektrum von CO2 nur zwei Banden beobachtet werden.
Die wichtigsten Schwingungsarten von Molekülen sind Streck- bzw. Valenzschwingungen (abgekürzt: ν) und Deformationsschwingungen (abgekürzt: δ). Daneben können vor allem nicht-lineare Moleküle eine Vielzahl weiterer Schwingungen ausführen, die man z.B. als "rocking", "twisting" oder "wagging" bezeichnet.
Instrumentelles
Zur Aufnahme von IR-Spektren können zwei prinzipiell unterschiedliche Spektrometertypen verwendet werden, wobei die älteren Scanning-Spektrometer mehr und mehr von den moderneren FT-IR-Spektrometern verdrängt werden. Auf der Vorlage zu Kapitel 2.2. sind die Funktionsweisen beider Spektrometertypen schematisch dargestellt. Gegenüber den Scanning-Spektrometern ermöglichen FT-IR-Spektrometer eine schnellere Messung, das Signal-Rausch-Verhältnis ist deutlich günstiger und die Wellenzahl-Präzision ist höher.
Bei Verwendung geeigneter Küvetten können von gasförmigen Proben oder von in einem geeigneten Lösungsmittel wie CCl4 gelösten Verbindungen IR-Spektren aufgenommen werden. Flüssige Proben werden häufig als dünner Film zwischen zwei NaCl Platten vermessen. Für feste Proben eignet sich eine analoge Aufnahmetechnik wenn man die Proben fein gemörsert in Öl suspendiert. Häufiger werden feste Proben allerdings mit einem Überschuss KBr fein verrieben und in einer hydraulischen Presse im Vakuum zu einem Pressling komprimiert. Dieser wird im Strahlengang des Spektrometers plaziert.
Das IR-Spektrum
Als Beispiel ist in der folgenden Abbildung das IR-Spektrum von 4-Methylbenzoesäureethylester abgebildet.
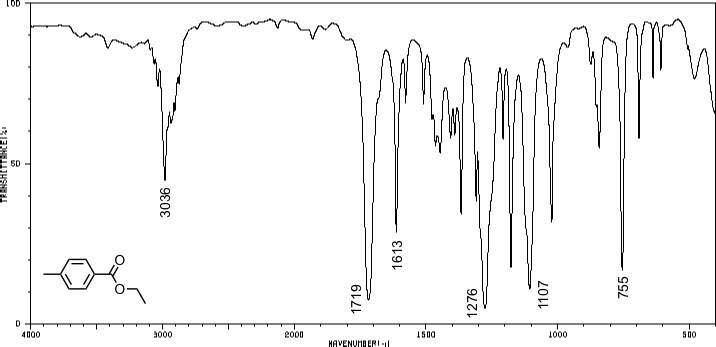
[Quelle: SDBS (Integrated Spectral Data Base System for Organic Compounds), National Institute of Advanced Industrial Science and Technology Japan]
Die markierten Banden in diesem Spektrum können folgenden Schwingungen zugeordnet werden.
| 1/λ / cm-1 | Zuordnung |
| 3036 | ν(C-H) Aryl (darunter ν(C-H) Alkyl) |
| 1719 | ν(C=O) Ester |
| 1613 | ν(C=C) |
| 1276 | ν(=C-O-C) |
| 1107 | ν(=C-O-C) |
| 755 | δ(C-H) des para-substituierten Aromaten |
Allgemein werden IR-Spektren in folgende Bereiche eingeteilt.
| 1/λ / cm-1 | Absorptionen |
| 3700 - 2500 | O-H, N-H, C-H Einfachbindungen |
| 2500 - 1900 | Dreifachbindungen und kumulierte Doppelbindungen |
| 1900 - 1500 | C=O, C=C, C=N, N=O Doppelbindungen |
| unterhalb 1500 | fingerprint-Bereich (Deformationsschwingungen, Valenzschwingungen von Gruppen mit schweren Atomen, Gerüstschwingungen) |
Gruppenfrequenzen
Als Gruppenfrequenzen bezeichnet man die charakteristischen Absorptionen eines Moleküls, die auf Schwingungen einzelner Strukturelemente zurückzuführen sind. Diese Schwingungen können immer dann beobachtet werden, wenn die Kopplung zu den Schwingungen des restlichen des Moleküls schwach ist. Da die meisten funktionellen Gruppen charakteristische Schwingungsfrequenzen besitzen, die sich deutlich von den Schwingungsfrequenzen anderer funktioneller Gruppen unterscheiden, geben das Auftreten und die genaue Lage der entsprechenden Absorptionsbanden in einem IR-Spektrum wichtige strukturelle Informationen. In den Vorlagen zu Kapitel 2.4. sind die wichtigsten Gruppenfrequenzen organischer Moleküle zusammengefasst.
Materialien
| zu Kapitel 2.1. | Harmonischer und anharmonischer Oszillator |
| zu Kapitel 2.1. | Schwingungsmoden von Methan |
| zu Kapitel 2.3. | IR-Spektrometer |
| zu Kapitel 2.4. | IR-Spektrum |
| zu Kapitel 2.5.1. | C-H Absorptionen |
| zu Kapitel 2.5.2. | O-H und N-H Absorptionen |
| zu Kapitel 2.5.3. | Absorptionen von Doppelbindungen |
| zu Kapitel 2.5.4. | Absorptionen aromatischer Verbindungen |
| zu Kapitel 2.5.5. | Absorptionen von Dreifachbindungen |
| zu Kapitel 2.5.6. | Absorptionen im fingerprint-Bereich |
Übungen
Spektreninterpretation
| 1. Übung | 1. Lösung |
| 2. Übung | 2. Lösung |
| 3. Übung | 3. Lösung |
| 4. Übung | 4. Lösung |
Aufgaben
- Ordnen Sie die Valenzschwingungen der Bindungen in folgenden Teilaufgaben jeweils in Richtung abnehmender Wellenzahl.
a) C,C-Einfachbindung, C,C-Doppelbindung, C,C-Dreifachbindung
b) C-H, C-D, C-C, C-Cl
c) C-H, O-H, N-H
Antwort - Ordnen Sie die C=O Valenzschwingungen der Verbindungen in folgenden Teilaufgaben jeweils in Richtung abnehmender Wellenzahl.
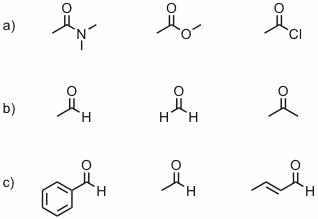
Antwort - Geben Sie an, welche der folgenden sechs Schwingungen IR-aktiv sind und welche Raman aktiv.
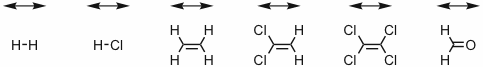
Antwort
Literatur
- H. Günzler, H.-U. Gremlich, IR-Spektroskopie, 4. Auflage, Wiley-VCH, Weinheim, 2003
- M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in der Organischen Chemie, 6. Auflage, Thieme Verlag, Stuttgart, 2002
- S. Wartewig, IR and Raman Spectroscopy, Wiley-VCH, Weinheim, 2003
- D. H. Williams, I. Fleming, K.-P. Zeller, Strukturaufklärung in der organischen Chemie, 6. Überarbeitete Auflage, Thieme Verlag, Stuttgart, 1991
Online-Datenbanken mit einer Vielzahl von IR-Spektren organischer Verbindungen:
Weitere Informationen zur IR-Spektroskopie finden sich unter folgenden Links:
Letzte Änderung: 23-03-30. Email